 Evolution
Evolution
On Population Genetics Estimates
 In his review of our book Science and Human Origins, Paul McBride wonders why I have not engaged the broader population genetics literature on human origins, but instead chose to focus on a single paper from 1995 by Francisco Ayala.
In his review of our book Science and Human Origins, Paul McBride wonders why I have not engaged the broader population genetics literature on human origins, but instead chose to focus on a single paper from 1995 by Francisco Ayala.
As I stated in the book, I chose that paper because in my opinion it presented the most difficult challenge to a very small bottleneck in our history as a species. If Ayala was right, and we shared 32 allelic lineages with chimps, then there was no way for a bottleneck as small as two individuals to have occurred. That kind of evidence, if substantiated, would have been conclusive.
That’s why I found it so fascinating as I watched his analysis crumble in the light of later research.
I was very aware that others besides Ayala have investigated human origins, using other methods and data. I chose not to address those studies directly in the book because I wanted to focus on the intriguing problem of HLA-DRB1’s patchwork phylogenetic history. I did allude to them in discussing problems with retrospective analyses, however. The fact that I had not addressed those alternate estimates is one reason why I never claimed to have proved the existence of a two-person bottleneck, but rather questioned the rush to judgment against such a bottleneck on the part of others.
So now, let’s consider how much these other methods add to the discussion.
With the advent of modern sequencing capabilities, much more data on human variability is now available than Ayala had access to. The HapMap and the 1000 Genomes projects, in combination with new analytic techniques, have both been used to try to establish dates of divergence and sizes of ancient human populations. It is now common to use non-coding sequences scattered across our genome for this kind of analysis, in an attempt to “average out” unusual gene-specific behavior and effects due to selection.
For example, a study published in 2011 used multiple genome sequences sampled from different chromosomes to investigate different models of human origins. The authors estimated the ages of different autosomal chromosomes as compared to the X, Y, and mitochondrial age estimates, and then used those estimates to examine different models for human history using Bayesian analysis.
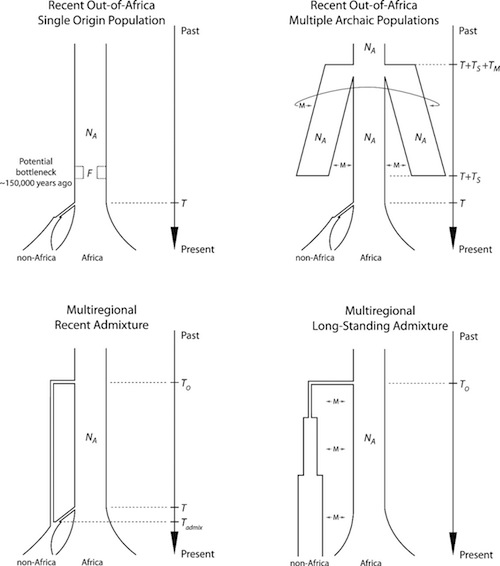
Their conclusion was that we could have had a single out-of-Africa origin, provided that the effective population size at the time of our origin was about 14,000, with a range of 12,000 to 17,000, for that particular population model. Other models gave other estimates, as can be seen below.
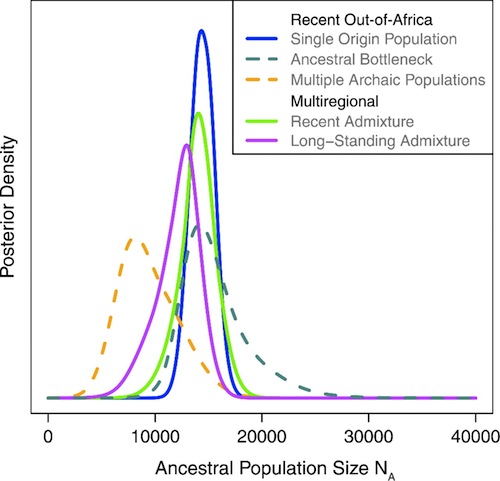
Estimates in that paper for the time to our most recent common ancestor (MRCA) with chimps also varied considerably, depending on which chromosomes were studied, which models of migration were used, and how many bottlenecks were proposed. They estimated the time of divergence of true humans from the primate lineage to be some time in the last 380,000 to 2,400,000 years. That’s a very big range. I like this study, though, because they are explicit about the imprecision of their estimates, and the effects of different historical scenarios on results.
Published estimates for ancestral population sizes vary from 100,000 to 1,000 — you can find some of them as references in the paper by Li and Durbin (2012) that McBride cited. Why such a large range of ancestral population sizes? First, the epoch chosen for study matters. The further back in time one goes, the more confounding factors can intervene. Population bottlenecks, changing selection, inbreeding, migratory behavior, strong selection for one gene accompanied by hitchhiking of neighboring genes — all these affect genome dynamics. Convergent evolution, parallel mutations, back-mutations, or gene conversion can obscure lineages and complicate tree-drawing. The method also matters. Linkage disequilibrium studies can’t go too far into the past because the recombination signal is either too small (too recent) or lost by repeated shuffling (too old), whereas allelic polymorphism studies, like Ayala’s, can go deep in evolutionary time.
But more worrying to me are the hidden assumptions in evolutionary models. Population genetics is a theory-laden subject, based entirely on neo-Darwinian assumptions. These assumptions, combined with over-simplifications required by current model building and/or mathematical analysis, can lead to erroneous claims about past genetic history.
Because of these difficulties, in my opinion it is an open question whether present genetic diversity provides sufficient information from which to draw conclusions about ancient populations. Determining events in deep human history may be beyond the reach of population genetics methods.