 Evolution
Evolution
 Intelligent Design
Intelligent Design
Exon Shuffling: Evaluating the Evidence
Yesterday, I introduced the subject of exon shuffling and gave a few reasons why such a mechanism cannot explain the origins of the earliest, most ancient proteins. Now I want to offer an evaluation of the arguments, pro and con, for the role of exon shuffling in accounting for the subsequent origins of novel protein folds.
The Case for Exon Shuffling
What, then, are the best arguments for exon shuffling? If the thesis is correct, a prediction would be that exon boundaries should correlate strongly with protein domains. In other words, one exon should code for a single protein domain. One argument, therefore, points to the fact that there is a statistically significant correlation between exon boundaries and protein domains (e.g., see Liu et al., 2005 and Liu and Grigoriev, 2004).
However, there are many, many examples where this correspondence does not hold. In many cases, single exons code for multiple domains. For instance, protocadhedrin genes typically involve large exons coding for multiple domains (Wu and Maniatis, 2000). In other cases, multiple exons are required to specify a single domain (e.g. see Ramasarma et al., 2012; or Buljan et al., 2010).
A further argument for the role of exon shuffling in protein evolution is the intron phase distributions found in the exons coding for protein domains in humans (for the significance of this, see my previous article). In 2002, Henrik Kaessmann and colleagues reported that “introns at the boundaries of domains show high excess of symmetrical phase combinations (i.e., 0-0, 1-1, and 2-2), whereas nonboundary introns show no excess symmetry” (Kaessmann, 2002). Their conclusion was thus that “exon shuffling has primarily involved rearrangement of structural and functional domains as a whole.” They also performed a similar analysis on the nematode worm Caenorhabditis elegans, finding that “Although the C. elegans data generally concur with the human patterns, we identified fewer intron-bounded domains in this organism, consistent with the lower complexity of C. elegans genes.”
Another line of evidence relates to genes that appear to be chimeras of parent genes. These are typically associated with signs indicative of its mode of origin. One famous example is the jingwei gene in Drosophila, which may have arisen when “the sequence of the processed Adh [alcohol dehydrogenase] messenger RNA became part of a new functional gene by capturing several upstream exons and introns of an unrelated gene” (Long and Langley, 1993).
We must take care, however, not to confuse the observed pattern of intron phase distribution, or exon/domain mapping, with proof that exon shuffling is actually the process by which this pattern arose. Perhaps common ancestry is the cause, but this must be demonstrated and not assumed. It is the biologist’s duty to determine whether unintelligent chance-based mechanisms actually can produce novel genes in this manner. It is to this question that I now turn.
The Problems with Domain Shuffling as an Explanation for Protein Folds
While the hypothesis of exon shuffling does, taken at face value, have some attractive elements, it suffers from a number of problems. For one thing, the model at its core presupposes the prior existence of protein domains. A protein’s lower-level secondary structures (α-helices and β-strands) exist stably only in the context of the tertiary structures in which they are found. In other words, the domain level is the lowest level at which self-contained stable structural modules exist. This leaves the origins of these domains in the first place unaccounted for. But stable and functional protein domains are demonstrably rare within amino-acid sequence space (e.g. Axe, 2010; Axe, 2004; Taylor et al., 2001; Keefe and Szostak, 2001; Reidhaar-Olson and Sauer, 1990; Salisbury, 1969).
A fairly recent study examined many different combinations of E. coli secondary structural elements (α-helices, β-strands and loops), assembling them “semirandomly into sequences comprised of as many as 800 amino acid residues” (Graziano et al., 2008). The researchers screened 108 variants for features that might suggest folded structure. They failed, however, to find any folded protein structures. Reporting on this study, Axe (2010) writes:
After a definitive demonstration that the most promising candidates were not properly folded, the authors concluded that “the selected clones should therefore not be viewed as ‘native-like’ proteins but rather ‘molten-globule-like'”, by which they mean that secondary structure is present only transiently flickering in and out of existence along a compact but mobile chain. This contrasts with native-like structure, where secondary structure is locked-in to form a well defined and stable tertiary fold. Their finding accords well with what we should expect in view of the above considerations. Indeed, it would be very puzzling if secondary structure were modular.
“For those elements to work as robust modules,” explains Axe, “their structure would have to be effectively context-independent, allowing them to be combined in any number of ways to form new folds.” In the case of protein secondary structure, however, this requirement is not met.
The model also seems to require that the diversity and disparity of functions carried out by proteins in the cell can in principle originate by mixing and matching prior existing domains. But this presupposes the ability of blind evolutionary processes to account for a specific “toolbox” of domains that can be recombined in various ways to yield new functions. This seems unlikely, especially in light of the estimation that “1000 to 7000 exons were needed to construct all proteins” (Dorit et al., 1990). In other words, a primordial toolkit of thousands of diverse protein domains needs to be constructed before the exon shuffling hypothesis even becomes a possibility. And even then there are severe problems.
A further issue relates to interface compatibility. The domain shuffling hypothesis in many cases requires the formation of new binding interfaces. Since amino acids that comprise polypeptide chains are distinguished from one another by the specificity of their side-chains, however, the binding interfaces that allow units of secondary structure (i.e. α-helices and β-strands) to come together to form elements of tertiary structure is dependent upon the specific sequence of amino acids. That is to say, it is non-generic in the sense that it is strictly dependent upon the particulars of the components. Domains that must bind and interact with one another can’t simply be pieced together like LEGO bricks.
In his 2010 paper in the journal BIO-Complexity Douglas Axe reports on an experiment conducted using β-lactamase enzymes which illustrates this difficulty (Axe, 2010). Take a look at the following figure, excerpted from the paper: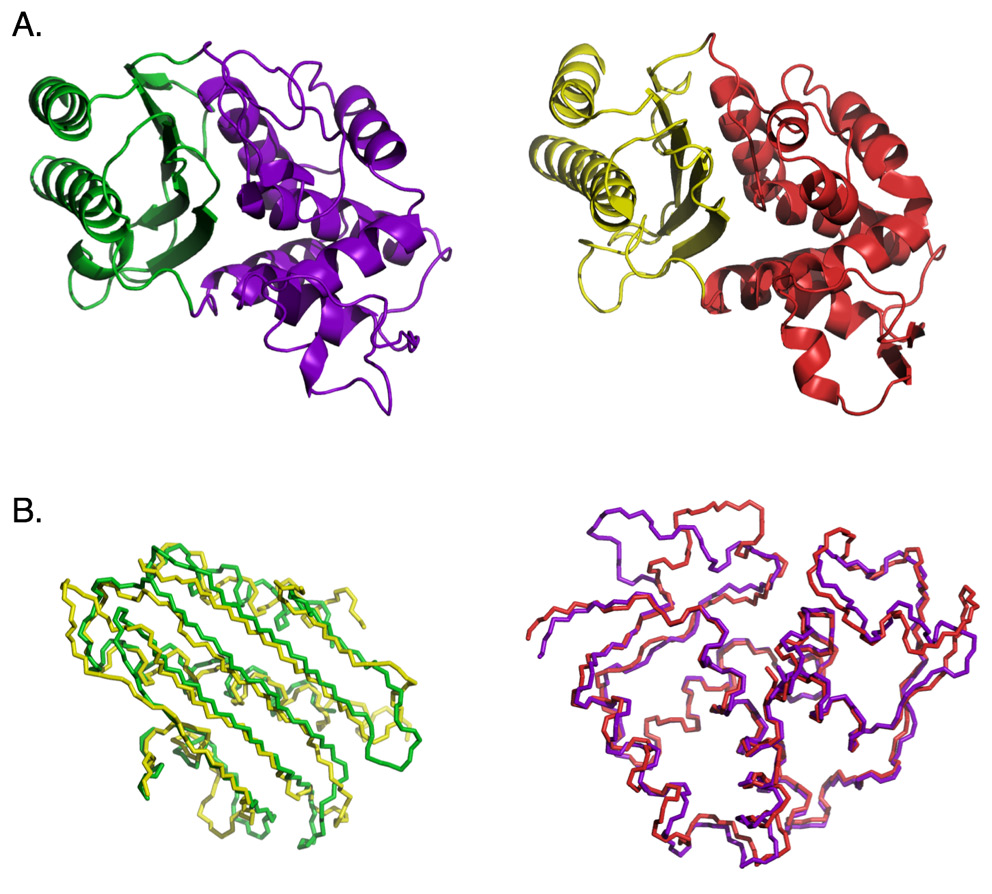
The top half of the figure (labeled “A”) reveals the ribbon structure of the TEM-1 β-lactamase (left) and the PER-1 β-lactamase (right). The bottom half of the figure (labeled “B”) reveals the backbone alignments for the two corresponding domains in the two proteins. Note the high level of structural similarity between the two enzymes. Axe attempted to recombine sections of the two genes to produce a chimeric protein from the domains colored green and red. Since the two parent enzymes exhibit extremely high levels of structural and functional similarity, this should be expected to work. No detectable function was identified in the chimeric construct, though, presumably as a consequence of the substantial dissimilarity between the respective amino-acid sequences and the interface incompatibility between the two domains.
This isn’t by any means the only study demonstrating the difficulty of shuffling domains to form new functional proteins. Another study by Axe (2000) described “a set of hybrid sequences” from “the 50%-identical TEM-1 and Proteus mirabilis β-lactamases,” which were created such that the “hybrids match[ed] the TEM-1 sequence except for a region at the C-terminal end, where they [were] random composites of the two parents.” The results? “All of these hybrids are biologically inactive.”
In fact, in the few cases where protein chimeras do possess detectable function, it only works for the precise reason that the researchers used an algorithm (developed by Meyer et al., 2006) to carefully select the sections of a protein structure that possess the fewest side-chain interactions with the rest of the fold, and chose parent proteins with relatively high sequence identity (Voigt et al., 2002). This only serves to underscore the problem. Even in the Voigt study, the success rate was quite low, even with highly favorable circumstances, with only one in five chimeras possessing discernible functionality.
Conclusion
To conclude, although there is some indirect inferential evidence for the role of exon shuffling in protein evolution, a consideration of how such a process might work in reality reveals that the hypothesis itself is fraught with severe difficulties.