 Evolution
Evolution
 Intelligent Design
Intelligent Design
Protein Evolution: A Guide for the Perplexed
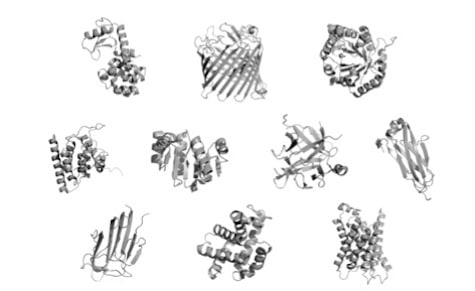
Proteins come in many shapes and sizes, as you can see in the illustration above taken from Doug Axe’s paper, “The Case Against a Darwinian Origin of Protein Folds.” Some can perform their functions as a single folded “domain,” a coherent, stably folded unit of protein structure. Others are composed of multiple linked domains, or even separate folded chains that must come together to form the functional unit that is useful to the cell.
How proteins manage to fold into the correct shape is an area of active study that others have discussed recently here. What I want to address is why the problem of protein evolution is such a big deal, and what the recent disagreement with Martin Poenie is about. Dr. Poenie, a University of Texas biologist, has critiqued Stephen Meyer’s new book, Darwin’s Doubt, leading to an exchange here at ENV among Poenie, Doug Axe, and Jonathan M.
 As Steve Meyer explained in his book, the problem is that the number of possible protein sequences that could exist is very large, occupying a very large potential sequence space, but the number of proteins that do exist is much smaller, and they are widely scattered across sequence space (perhaps — in fact, that is one of the things being debated). The potential space is so large that a purely random search for rare functional proteins would spectacularly fail. So unless functional sequences are easy to find (very common), and/or are clustered together (easily reachable from one functional island to another), explaining current protein diversity without design is impossible.
As Steve Meyer explained in his book, the problem is that the number of possible protein sequences that could exist is very large, occupying a very large potential sequence space, but the number of proteins that do exist is much smaller, and they are widely scattered across sequence space (perhaps — in fact, that is one of the things being debated). The potential space is so large that a purely random search for rare functional proteins would spectacularly fail. So unless functional sequences are easy to find (very common), and/or are clustered together (easily reachable from one functional island to another), explaining current protein diversity without design is impossible.
Our interlocutors are keenly aware of the problem. To solve it, some propose that the first proteins were composed of just a few kinds of amino acids. Or that the first proteins were very small, or that they were very non-specific (“promiscuous” is the word used for this in the literature). This reduces the scope of the problem somewhat.
Others like to suggest that, having somehow stumbled on one or a few successful folds, evolution was able to bootstrap its way forward by a combination of gene duplication and cooption of the duplicates to new functions, or by recombining existing proteins into new functional forms. There is now also the suggestion that completely new proteins can sometimes be generated by the random insertion of mobile genetic elements into non-coding DNA, though this was thought to be very unlikely just a few years ago.
The problem with these scenarios is that they require proteins to be remarkably tolerant of sequence rearrangements and insertions, or remarkably easy to shift to new functions, or remarkably different at the dawn of life from what they are now — large, complex, macromolecules tailored for specific functions.
Given that no one knows how the chemistry of life could be carried out by a handful of non-specific proteins, most people opt for explanations having to do with recruitment of duplicate proteins to new functions by point mutation or rearrangement, or the de novo creation of new protein-coding genes from previously non-coding DNA.
Here is where it gets interesting. If you examine the arguments being made here, they appear to be self-refuting:
1. Proteins can be recruited to new functions, but only if you start with the right ancestral form. This is because multiple chemical interactions are required to give a protein its shape and catalytic activity; proteins that differ too much in sequence can have completely different networks of interactions, even if they share the same shape. This means that there may be no stepwise path to convert one to the other. Proteins are finicky things.
2. Introducing a few mutations can so disrupt an enzyme that its delicate catalytic activity is destroyed, making it impossible to recruit enzymes to new functions that are not already very similar in sequence, and/or already share some level of function. Once again, proteins are finicky things.
OR
3. Proteins are robust, and can easily be improved by recombining them. In this way, new combinations of mutations can be produced in one fell swoop, sidestepping potential non-functional combinations. (I’ll ignore the fact that recombination of this sort works only in diploid organisms, which leaves out bacteria and Archaea, and all the most ancient enzymes).
4.You can create a new functional gene by inserting whole chunks of DNA into prior non-coding DNA, that by blind luck, not design, is transcribed and translated into a new functional protein (something the organism actually benefits from).
So which is it? Hard or easy? Remember, for evolution to work, proteins need to be remarkably tolerant of sequence rearrangements, or remarkably easy to shift to new functions by amino acid substitutions, or functional sequences need to be quite common.
It would appear that #1 and #2 say the evolution of proteins is hard to explain. We agree. If it were easy to shift proteins to new functions, then something like the transition between kbl and bioF (published here) should have been possible. But claiming that the reason we failed was because we didn’t start with exactly the right ancestral form makes life’s history one long providential journey, or the product of an incredibly lucky series of accidents.
The paper by Romero and Arnold that Poenie cites in support of claim #3 is based on experiments that have nothing to do with the problem of Darwinian evolution, and everything to do with genetic engineering. The researchers sought to optimize already existing enzymes by recombining family members with the same structure and function, but different amino acid sequences. To ensure the greatest likelihood of success, the experimenters used an algorithm called SCHEMA to carefully choose recombination breakpoints at positions most likely to minimize side chain disruptions. So this experiment says nothing about what random recombination can do, or its ability to generate new function. In fact they clearly state that random recombination fails miserably by comparison.
Claim #4 is based on the fact that novel protein-coding sequences (orphan genes discussed here) appear to exist in our own genomes and elsewhere. But just to point out what should be obvious, the fact that something exists does not explain how it came to exist. Unless it can be shown that insertion of elements into random non-coding sequence in an unguided fashion really does produce functional proteins, then we can talk about it all we want but we still don’t know how they got there.
Finally Doug Axe and others have had something to say about the rarity of functional folds. His 2004 paper, “Estimating the Prevalence of Protein Sequences Adopting Functional Enzyme Folds,” came up with a shockingly small number. From the abstract:
Starting with a weakly functional sequence carrying [the pattern of hydropathic constraints along chains that form the beta-lactamase domain fold], clusters of ten side-chains within the fold are replaced randomly … and tested for function. The prevalence of low-level function in four such experiments indicates that roughly one in 1064 signature-consistent sequences [having the same hydropathic constraints] forms a working domain.
That number is based on experiment with real enzymes, not in silico mock-ups.
So which is it? From the above, proteins are not tolerant of sequence rearrangements, or easy to shift to new functions by amino acid substitutions, and functional sequences are quite rare. It would appear that the unguided evolution of proteins is hard, very hard.