 Evolution
Evolution
 Intelligent Design
Intelligent Design
Protein Folding: A Materialist’s Nightmare
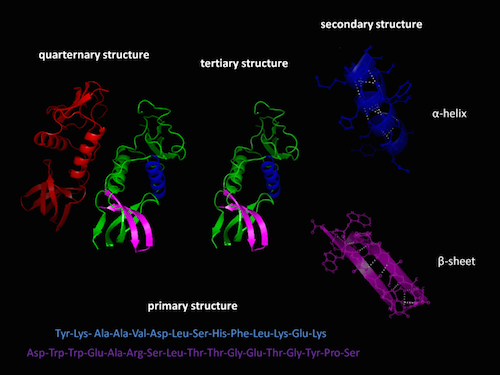
It’s one of the most astonishing phenomena in nature: a linear sequence self-organizes into a 3-D machine. Try arranging magnets on a string that can fold the string into a working wrench. That’s about the level of complexity that happens every day in every cell of your body: gene sequences translate into amino acid sequences that spontaneously fold into functional proteins. How do they do it?
PNAS published a "Perspective" article, "The Nature of Protein Folding Pathways," by S. Walter Englander and Leland Mayne. Unsurprisingly, they try to approach the problem from purely materialistic presuppositions. There is no mention of specificity, amino acid sequence, or digital information.
Knowing as they do that the space of possible folds is too vast for blind search, their entire discussion revolves around possible ways to break the folding problem into smaller problems that might be tractable to chance and the laws of thermodynamics. A bottom-up solution might involve smaller folds called "foldons" that combine through a sequence of steps down a "funnel-shaped energy landscape" that guides it toward the native fold. Does this provide a deterministic explanation?
A successful folding model must resolve major questions concerning folding time and energy. Levinthal pointed out that the vast array of protein conformations in unfolded space cannot simply reequilibrate and reach the unique native state by an undirected random search in any reasonable time. Early theoretical work therefore focused on the downhill energetic drive and the many independent routes that heterogeneity and microscopic thermal searching alone seemed to require. The new view answer to the "why" question is that, from the microscopic point of view, there seems to be no other viable choice.
Experimental work recounted here reveals an emergent macroscopic behavior that provides a previously unrecognized mechanism. Random search does not have to carry the protein all of the way to the native state. It only needs to accomplish the formation of a first native-like foldon. This process is thermodynamically downhill and is guided by the inherent cooperativity of native foldon units. (Emphasis added.)
While it is true that sophisticated techniques can see the protein fold as a series of stages, this ignores the main problem: How does DNA know in advance what sequence will build the first foldon, and the second, and the third, and so on, till a functional protein results?
The time scale for forming a first foldon unit by an unguided search, perhaps two segments ?20 residues in length, is shorter by far than for a reference 100-residue protein [3100/(2 � 320) ? 1040]. The formation of subsequent foldons must proceed by way of similar microscopic searching but in a more guided way analogous to the process of "folding upon binding." The concept that proteins start folding by forming a nativelike structural nucleus has been widely accepted. This minimal structure can be sufficient to seed subsequent foldon-foldon interaction steps in a sequence of more guided searches that follow through, rapidly, to the native target.
Their explanation is as senseless as suggesting that the unguided origin of a meaningful paragraph can be simplified by breaking the problem down into self-organizing words that spontaneously combine into the final paragraph.
The Levinthal space of conformations is no less vast if you try to search it blindly in parts. How does a DNA sequence become a final, functional whole? How does the amino acid translation know how to build the "nativelike structural nucleus" to "seed" the subsequent foldons? How did the gene plan subsequent foldons that would know where and when to interact with the structural nucleus? The authors are "helping themselves" to the notion that all this information can be extracted from the structure of the first foldon.
Englander and Mayne certainly know what they’re up against, trying to account for a functional fold by unguided processes. Nevertheless, they make quite a leap in assuming this problem can be solved by dividing it up:
A single residue has very low probability for finding its correct native partners in a sea of nonnative alternatives. Certainly, microscopic thermal searching must underlie any structure formation process. However, given the required energy bias computed by Zwanzig et al., it seems that microscopic-level searching alone cannot swiftly reach the native state. By contrast, in a more macroscopic foldon- based scenario each correct native-like choice is driven by the collective energy of many interaction sites held stereochemically in a native-like geometry in partner foldons.
You cannot solve an impossible blind search by dividing it up into smaller searches. If each small search is unguided, the whole search is unguided. To remain materialists, they have to assume that "there is no other viable choice":
The disordered microscopic multitrack search envisioned in the paradigmatic new view model describes the initial stage amino acid-level search to form cooperative nativelike foldon structures, but not the final native state. Experiment displays an emergent foldon-based macroscopic behavior that provides the structural guidance and free energy bias for the ordered stepwise formation of discrete native-like intermediates in a folding pathway that leads to the native state.
Folding in moderately small, separately cooperative units may be necessary for proteins to fold at all. A much larger step size would confront the Levinthal time scale problem; much smaller steps cannot assemble the energy bias required by the Zwanzig criterion for fast folding. Thus, as before for the microscopic view, it may be that there is no other viable choice.
Yes, there is another viable choice. It’s intelligent design.
The information in DNA cuts through the vast conformational search space, and finds the route to the functional fold rapidly, sometimes within microseconds. ID is the only way to navigate a vast search space and arrive at a destination on time. Intelligent design can build self-organizing systems that obey the laws of thermodynamics, just as in the analogy of magnets on a string spontaneously folding into a wrench. Even if entropy increases as the protein folds, guiding the polypeptide through the funnel-shaped energy landscape to the final product, this does not negate the design that went into the system. It amplifies it.
We would like to ask these authors how DNA knew to code for chaperones that can help other proteins fold correctly. Those chaperones had to be coded in DNA such that they would fold correctly themselves first. And add another question: how were all the other molecular machines in the cell built that repair or dispatch misfolded proteins? What created the Golgi apparatus where proteins are finished, packaged, and delivered to their work sites?
Okay, let’s leave it there. There’s no need to pile on the difficulties for materialism.
{kind=link}