 Evolution
Evolution
 Intelligent Design
Intelligent Design
More Secret Codes in “Junk DNA”
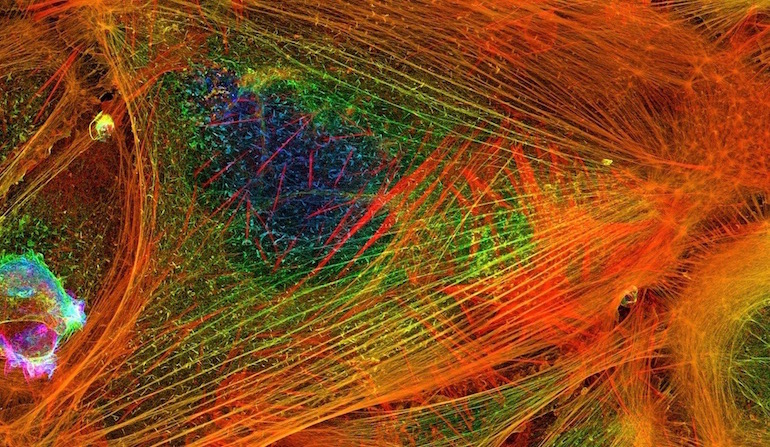
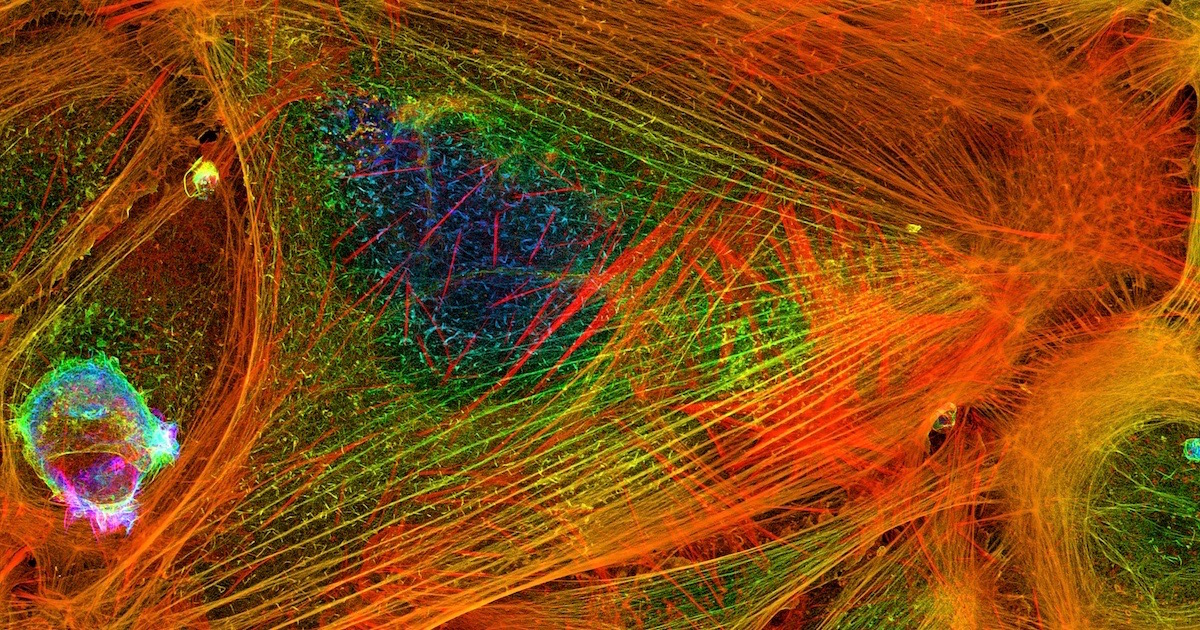
Scientists find the most interesting things when they suspect function in poorly understood parts of the genome, rather than relegating them to the junk pile as useless. Here are two recent examples.
Silent Code in Action in Actin
“Actin is an essential and abundant intracellular protein that plays a major role in developmental morphogenesis, muscle contraction, cell migration, and cellular homeostasis,” say Vedula et al. in a paper in the journal eLife. A protein this vital commands our attention. How does it perform so many different functions? What governs the destination and activity of the different forms of actin?
The paper reads like a scientific detective story. A team of researchers from the University of Pennsylvania and the National Institutes of Health wanted to know why two forms of actin (isoforms) are nearly indistinguishable in terms of their sequence (except for four amino acids at one end), but perform very different functions in the cell. They also found it intriguing that these isoforms, β-actin and γ-actin, are coded by different genes, but end up looking very similar.
Let’s divulge the conclusion in the title of the paper: “Diverse functions of homologous actin isoforms are defined by their nucleotide, rather than their amino acid sequence.” Do you hear the word “code” coming? How about “silent code”?
Here we tested the hypothesis that β- and γ-actin functions are defined by their nucleotide, rather than their amino acid sequence, using targeted editing of the mouse genome. Although previous studies have shown that disruption of β-actin gene critically impacts cell migration and mouse embryogenesis, we demonstrate here that generation of a mouse lacking β-actin protein by editing β-actin gene to encode γ-actin protein, and vice versa, does not affect cell migration and/or organism survival. Our data suggest that the essential in vivo function of β-actin is provided by the gene sequence independent of the encoded protein isoform. We propose that this regulation constitutes a global ‘silent code’ mechanism that controls the functional diversity of protein isoforms. [Emphasis added.]
Good old controlled experimentation, using the CRISPR editing tool, showed that editing the gene for one form produced working copies of the other form. Mice that had defective genes for γ-actin could be rescued by editing the β-actin gene to produce γ-actin. All they had to do was edit five nucleotides to produce healthy mice with no β-actin at all, even though previous knockout experiments showed that mice without the β-actin gene die early in development. How could this be?
Further experiments suggested that it’s not the resulting amino acid sequence that determines the function, but “silent” substitutions in the gene. Something in the β-actin gene was regulating the outcome in a different way, even though it generated only γ-actin. The γ-actin isoform went to where β-actin normally went, and performed its function as if it were β-actin.
The researchers note that different isoforms of actin can have vastly different ribosome densities, differing up to a thousand-fold. In the cytoplasm, some isoforms can compensate for other ones. This arrangement provides flexibility to the cell in most cases:
These results suggest the actin isoform with similar ribosome density can plausibly compensate for the loss of one of the isoforms. In agreement, given the orders of magnitude difference in ribosome density between β-actin and other actin isoforms, none of the other actin isoforms can compensate for the loss of β-actin. We propose that changes in ribosome density arising from silent substitutions in nucleotide sequence, affect translation dynamics and protein accumulation rates, which in turn regulate functional diversity of actins.
The authors feel this kind of “silent code” may be at work in other protein families as well. The word “code” is ubiquitous throughout this paper. In another case, they describe the targeting of one actin isoform to the cell periphery by what they call “zipcode-mediated transport.” They have more to say about coding than evolution, in fact, except in one paragraph where they invoke the common Darwinian excuse that an essential gene tends to be conserved against alteration:
Despite the fact that non-muscle actin isoform genes have evolutionarily diverged > 100 million years ago, they have retained remarkable sequence conservation, far higher than what would be expected if the synonymous substitutions in their coding sequence were completely randomized. (Erba et al., 1986). This is consistent with our idea that actin isoform coding sequence exists under additional evolutionary pressure, over and above the conservation of amino acid sequence. We propose that at least some of this pressure is aimed to maintain the divergent translation dynamics within the actin family, in order to drive their divergent functions.
It appears, however, that intelligent design research could be more productive in follow-up studies. They conclude, “Further systematic analysis of knockouts of homologous isoforms would enable establishing the universality of the ‘silent code.’”
Dark Matter in Your Brain
A more appropriate term for “junk DNA” might be “dark matter” — sequences that are not yet understood. Nature News illustrates a good use of this metaphor in an article, “‘Dark matter’ DNA influences brain development.” Amy Maxmen writes, “Researchers are finally figuring out the purpose behind some genome sequences that are nearly identical across vertebrates.”
A puzzle posed by segments of ‘dark matter’ in genomes — long, winding strands of DNA with no obvious functions — has teased scientists for more than a decade. Now, a team has finally solved the riddle.
The conundrum has centred on DNA sequences that do not encode proteins, and yet remain identical across a broad range of animals. By deleting some of these ‘ultraconserved elements’, researchers have found that these sequences guide brain development by fine-tuning the expression of protein-coding genes.
There’s no reason to suspect that any of the heroes of this article doubt evolutionary theory. But one lead researcher of a new paper did what a good design scientist would do: keep looking for function until you find it.
The results, published on 18 January in Cell, validate the hypotheses of scientists who have speculated that all ultraconserved elements are vital to life — despite the fact that researchers knew very little about their functions.
“People told us we should have waited to publish until we knew what they did. Now I’m like, dude, it took 14 years to figure this out,” says Gill Bejerano, a genomicist at Stanford University in California, who described ultraconserved elements in 2004.
What they found is the opposite of evolutionary expectations, even though the article assumes evolution:
Bejerano and his colleagues originally noticed ultraconserved elements when they compared the human genome to those of mice, rats and chickens, and found 481 stretches of DNA that were incredibly similar across the species. That was surprising, because DNA mutates from generation to generation — and these animal lineages have been evolving independently for up to 200 million years.
Genes that encode proteins tend to have relatively few mutations because if those changes disrupt the corresponding protein and the animal dies before reproducing, the mutated gene isn’t passed down to offspring. On the basis of this logic, some genomicists suspected that natural selection had similarly weeded out mutations in ultraconserved regions. Even though the sequences do not encode proteins, they thought, their functions must be so vital that they cannot tolerate imperfection.
You have to wonder what function Darwinian evolution had in this research. The expectations were wrong, the results were surprising, and the team found more design than was previously known — to the point of implying perfection. The only evolution-talk sounds like an after-the-fact gloss to keep the preferred narrative from being falsified.
For more on the tortured subject of supposed trash in the genome, see The Myth of Junk DNA, by Jonathan Wells.
Photo: Actin filaments, by Howard Vindin (Own work) [CC BY-SA 4.0], via Wikimedia Commons.
{kind=link}