 Evolution
Evolution
 Intelligent Design
Intelligent Design
Why Their Separate Ancestry Model Is “Wildly Unrealistic”
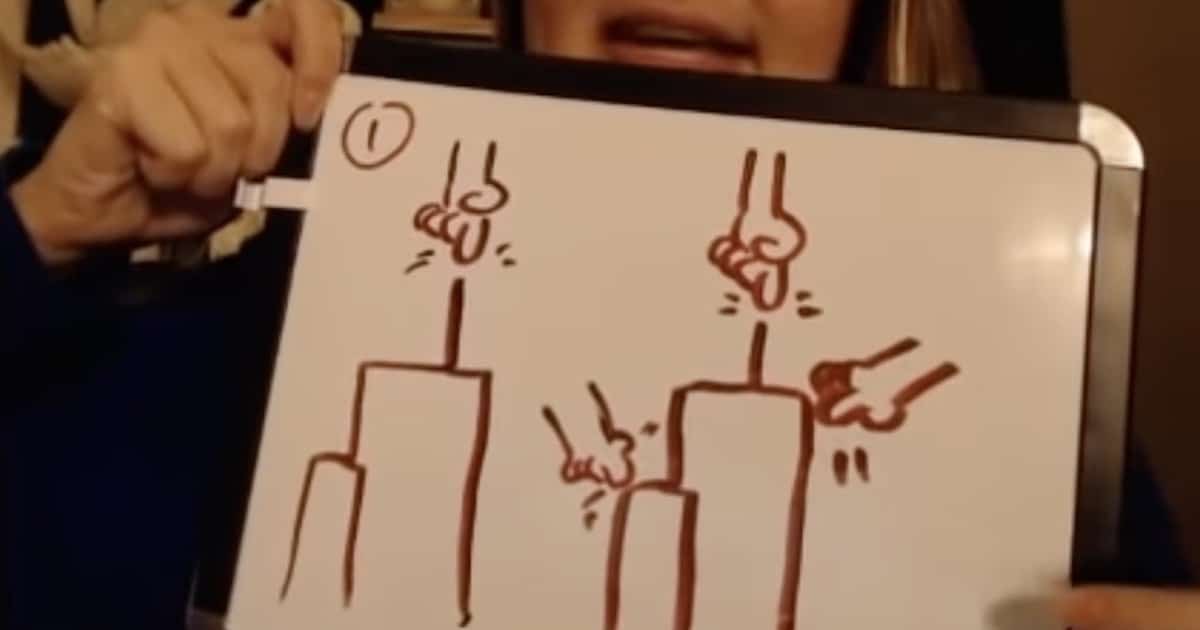
In my post yesterday I outlined how Erika, aka the popular evolution YouTuber “Gutsick Gibbon,” critiqued my earlier post, which commented on an important paper in the field of phylogenetics, Baum et al. (2016), which purported to test separate ancestry. Between 7:55 and 9:24 of her response video, Erika shows a diagram (above) to respond to my point that the Baum et al. (2016) paper tested a model of separate ancestry that is not endorsed by anyone in the ID community.
Here’s what Erika, aka Gutsick Gibbon, is saying. In the diagram, she has two different models of what creationists (left) and intelligent design (ID) proponents (right) might be saying. (Note that she disagrees with both; she’s just trying to describe what she thinks the groups are saying.) Each “finger” in the diagram is supposed to represent an instance where the designer acted to influence the course of biological history. The left tree is supposed to show what she thinks the creationist’s model is that was tested by Baum et al. (2016). She mistakenly thinks that ID proponents are angry because we’re really putting forth some model like the diagram on the right — where a designer creates a group but then allows evolutionary tinkering. So, she thinks we’re upset because Baum et al. (2016) didn’t include tinkering in their model. This is actually not the case.
When I argued that Baum et al. (2016) failed to properly test separate ancestry, that has nothing to do with a failure to incorporate “tinkering” into the model. Also, as a side note, ID proponents do not advocate a “tinkering” hypothesis. This is a common misconception about the ID view. Instead, the primary objection of ID proponents to the Baum et al. (2016) paper is due to how the separate ancestry and family ancestry models were created in the first place. In short, Baum et al. (2016) assumes that shuffling of the synapomorphies is an accurate model for separate ancestry. ID proponents and others who have a design-based perspective would heartily reject that, for reasons I will explain.
How They Chose Data for the Separate Ancestry Model
Baum et al. (2016) uses several different datasets to test separate ancestry. Their molecular dataset, our focus here, is from a 2011 paper by Perelman et al. where 54 genes were used to construct a molecular phylogeny of living primates. (We’ll call this the “Perelman dataset.”) Primates all have around 30,000 genes, so the first question is how the authors got from 30,000 genes to 54. Note the details given from Perelman et al. (2011):
A complete list of 54 primer sets used in this study is presented in Table S2. This list includes primers from earlier studies (Murphy et al. 2001), as well as those designed specifically for this study using a unique bioinformatics approach (Pontius, unpublished data). (Perelman et al. 2011)
If you look at Table S2 in you can see that the majority (38 of the 54 genes) came either from Murphy et al. (2001) (9 genes) or were specially designed for the study and no details are given (29 genes). For our purpose, let’s just look at the 9 taken from Murphy et al. (2001) which are described as being selected in the following way:
The GenBank and UniGene databases (NCBI) were searched for genes with exons of sufficient length (>200 bp) and variability (80±95% nucleotide identity between mouse and human), thereby providing adequate variation for the purpose of phylogenetic and somatic cell/ radiation hybrid mapping. (Murphy et al. 2001)
What’s Happening Here?
Two types of selection or filtering are going on when they choose genes for their study. The first selection is to remove genes not present in all the species they were studying. In other words the genes had to be within the databases, be of sufficient size, and exist in all the species being considered. This rules out species-specific genes such as orphan genes. The second selection is that genes having the greatest number of phylogenetically informative sites or synapomorphies were chosen. (A synapomorphy is a variant/trait that is shared by at least two descendent taxa and thought to be inherited from a most recent common ancestor, where it evolved.) In order to win at this second selection, a gene should have the greatest number of variants which differ between at least two taxa, but in the same way (i.e., at site 1 two taxa have ‘A’s while the ancestral site was a ‘G’). A simpler way of putting this is they picked genes that varied the most between species, but in a likely functional way. If you look at Table 1 from Murphy et al (2001) you can see that the ADORA3 gene has 191 phylogenetically informative sites out of 330 base pairs. That means that at 191 positions it differs between at least two of the comparison taxa in a consistent way. To help better understand why these filters are stacking the deck from a design perspective, I want to give an analogy.
Let’s do a thought experiment where we want to create a distance tree demonstrating the evolution of household chairs. We choose five traits to compare. Those five traits must be exhibited by all the chairs and the five traits must vary between at least two chairs. This type of selection effectively eliminates unique properties of the chairs such as leather because not all chairs have this. Instead it prioritizes specific parts of chairs that were made intentionally different for functional or economic reasons. This type of selection on a designed object, such as a chair, will create an intuitive hierarchical tree even though the chairs are not actually related. How? Read on.
For example, screws, which hold chairs together, are likely to be a trait shared by all chairs (first criterion passed), but screws aren’t likely to vary a lot between chairs and therefore wouldn’t be selected as a trait. On the other hand, legs are a trait shared by all chairs and these are likely to vary quite a bit between different chairs based on the function of the chair. Children’s chairs will have shorter legs. Decorative chairs will have aesthetically pleasing legs. Folding chairs will have collapsible legs. Some chairs may have four legs while others five or even more. Seats are another example of a trait that will be common to all but differ a lot. Children’s seats on chairs will be smaller. Decorative chairs will have aesthetically pleasing seats. Collapsible chairs will have mobile seats. Thus, a selection for “differentness” with designed objects enriches for traits that cluster due to functional constraints or compatibility, not ancestry. If organisms are in fact designed, a very similar phenomena could be occurring in these phylogenetic comparisons.
Now let’s look at how this data set was used by Baum et al. (2016) in their separate ancestry model and what about the model is so problematic from an ID perspective.
They Used Synapomorphy Shuffling to Test Separate Ancestry
In describing the separate ancestry model Baum et al. (2016) says:
A key feature of the species SA [separate ancestry] model is that for each character [meaning genetic variants or fossil characters] the state drawn by each species is independent of that drawn by other species.
But what do they mean by saying that the state drawn by each species is “independent” of that drawn by another species? How are they actually creating their separate ancestry model? What I’ve gathered is this essentially means that in their “separate ancestry” model, the traits or synapomorphies were shuffled randomly to create a hypothetical model of what they think separate ancestry would be. I will illustrate with an example in Figure 1 adapted from the molecular Perelman dataset, where I took actual names of genes used by Baum et al. (2016), but then represented the different synapomorphies of those genes as spelling changes for the function of the gene.
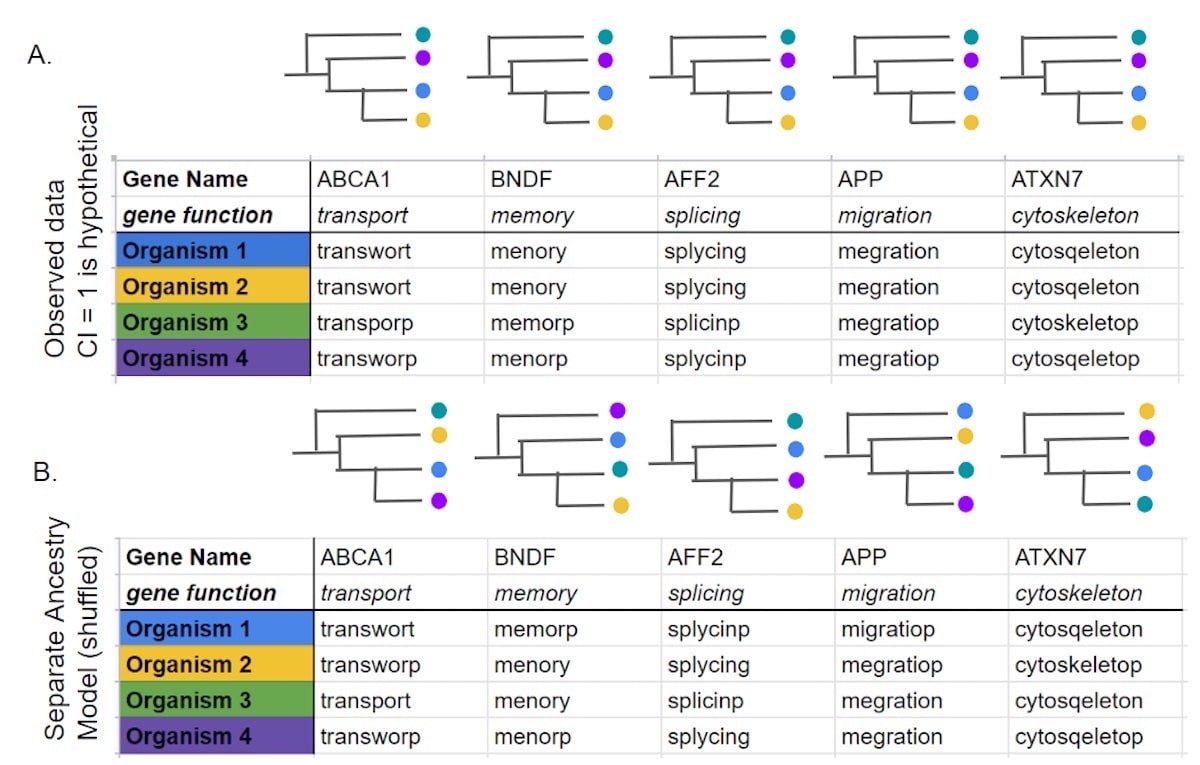
To elaborate, in Figure 1 above are genes ABCA1, BNDF, AFF2, APP, and ATXN7. I have represented their DNA sequences simply as lowercase letters (transport, memory, splicing, migration, and cytoskeleton respectively) corresponding to their major functions. Then, to represent the synapomorphies between these organisms I’ve introduced some spelling errors. For example, in Figure 1A the ABCA1 gene in organism 1 is the sequence is transwort (lowercase) and BNDF is menory, AFF2 is splycing, APP is megration, and ATXN7 is cytosqeleton respectively. The pattern of changes within the gene (columns) are the same for all five genes in Figure 1A — notice how top to bottom the phylogenetic trees are the same. Thus, Figure 1A represents the data observed with one important caveat — I have artificially made the pattern of synapomorphies perfect (CI =1) just for clarity (not the case in the real data).
Now, the authors Baum et al. (2016) constructed their separate ancestry model by permuting (shuffling) the synapomorphies of these sequences (Figure 1B), in a random manner which assumed there would be no reason to find correlations of traits across different organisms. Here’s how they described their methods:
To evaluate whether the observed hierarchical signal is more than expected under species of family SA, we used the PTP test which uses a Monte Carlo approach to simulate data under the SA hypothesis. We implemented PTP tests using the permute function of PAUP* ver. 4.0a134-146 with parsimony as optimality criterion and hence tree length as a measure of tree-like structure.
In other words, Baum et al. (2016) gave the synapomorphy of the ABCA1 gene sequence of organism 2 to organism 4 and vice versa using a permute function (see Figure 1B). They did not just swap the whole genome sequence between two organisms but swapped individual characters — in this case base pairs — to remove the connection between them (notice how top to bottom the colors are scrambled). As expected, after random shuffling of the synapomorphies the tree length drastically increased — meaning more evolutionary events were required to explain the data, and the tree was not very parsimonious (see Table 1 from Baum et al. (2016)).
Following this they calculated the p-values (see Table 3 from Baum et al. (2016)). The p-values are outrageously low, dictating a strong rejection of the separate ancestry model tested. Erika interprets this here as indicating that at least this model of separate ancestry is a totally unreasonable hypothesis. Their method is given below:
For many of the tests the observed test statistic fell well outside the range of values obtained under the SA hypothesis. In such cases, we report the distance of the observed data from the mean of the SA distribution in units of the SD (the z-score) and also provide a P-value assuming a normal distribution. Although the latter is only an approximation, it will provide the reader with a sense of how improbable the data would be under SA.
Of course, I reject this model of “separate ancestry” as well. This model, as they’ve developed it, is totally unrealistic for all kinds of reasons and therefore far less likely than the observed model. Now, anytime one observes this type of data in biology it should definitely make one question if not immediately reject the model. Erika also partially recognizes this because in reference to these zeros she also says “That’s insane! You don’t see that in regular science.” Basically, p-values this low typically indicate that there is something really wrong with one’s model. So, let’s talk about this, and what might be wrong with their model of separate ancestry.
Synapomorphy Shuffling is Not a Good Test of Separate Ancestry
The short(er) explanation for why synapomorphy shuffling is not a good model of separate ancestry is that synapomorphies or traits may cluster for designed systems based upon functional reasons like optimization, constraints, or compatibility. Recall that a synapomorphy is a trait or site uniquely shared by members of a group that helps to define that group. Under typical phylogenetic thinking, synapomorphies are thought to exist because they evolved in the common ancestor that gave rise to the group. But in an ID-based world, synapomorphies might exist because they represent a suite of traits required for a group of organisms to perform some important function related to their survival.
In designed systems, traits don’t vary randomly, and often vary according to predictable patterns which may be related to functional needs. In biology, these functional needs could be related to an organism’s niche, lifestyle, locomotion, metabolism, diet, or other behaviors. In other words, organisms which live in similar niches and/or have similar lifestyles, modes of locomotion, metabolisms, diets, or other behaviors, may tend to have similar traits all related to functional constraints that are required for that organism to survive in its environment. Thus, in a designed biosphere, traits won’t vary randomly but will follow similar patterns, correlations, and relationships across organisms according to their various survival needs. To put it simply, organisms with similar lifestyles will show similar architecture. This will be true not because of common ancestry but because of design constraints which must be fulfilled for an organism to survive in its environment.
Now, the Long(er) Answer…
ID proponents have a problem with this model of separate ancestry because it does not account for anticipated taxa-specific design constraints (aka what I am calling “functional synapomorphies”). Most ID proponents would hold that only synapomorphies that are historical in nature could be shuffled in such a fashion. As a thought experiment, if one selected synapomorphic chair traits, a very nice nested hierarchical pattern will result. Collapsible chairs will cluster, desk chairs will cluster, armchairs will cluster, children’s chairs will cluster, and the universal common ancestor might be something like a stool. Thus, if a synapomorphy is functional (i.e., contributes to the function) and not historical, this random shuffling of synapomorphies would be analogous to taking chair-specific design differences (like a collapsible seat and short legs for a children’s chair), mixing them up, and then observing that collapsible chairs and children’s chairs no longer group together. When you shuffle functional traits of designed objects, you will get statistical zeros, because you have obliterated the design signal. Most likely you’d also get some quite weird designs that don’t work very well! Imagine outdoor patio furniture with traits of indoor office chairs. It wouldn’t work!
Given how the data were selected in the first place, it is very likely that many of these synapomorphies are functional.
The reason why functional synapomorphies cannot be used is because hierarchical clustering of functional synapomorphies or traits are abundant in scenarios that we know have not arisen due to a process of descent with modification. Don’t like my chair analogy? Take the distance tree, created by Doolittle and Bapteste 2007, of French departments based on the number of shared sur-names (See Figure 1b in the paper). This is a great example of how functional synapomorphies or traits can result in logical clustering of data when no descent with modification process has occurred. Baum et al. (2016)’s error is therefore as follows: They assume that design must produce random distributions of traits. However, all of our experience with sets of designed systems shows this is not the case. Erika doesn’t appreciate this point, and thus she misunderstands our critique of the Baum et al. paper.
What we know about design, from engineering and other life scenarios, is that design often creates a hierarchical similarity pattern centered around function that could look like ancestry if one forces it. Why do designers produce these hierarchical patterns? They aren’t trying to be deceptive, mimicking systems that look like they are the product of common ancestry. Rather, designers are simply applying logical design considerations like optimization, constraints, compatibility, dependencies, or reuse during the design process.
Thus, I hold that the model of separate ancestry rejected in the Baum et al. (2016) paper is not endorsed by most in the ID community because it does not account for the design expectation that functional synapomorphies or traits will cluster due to optimization, constraints, and a need for compatibility.
On Monday, I will look at the consistency of the phylogenetically informative sites for the Baum et al. (2016) paper. Spoiler alert: It looks like design.