 Evolution
Evolution
On Retrospective Analysis and Coalescent Theory

Population genetics as a scientific discipline makes use of mathematics and the principles of neo-Darwinism to try to understand how genetic variation spreads through populations and influences their evolution. It does so by assuming that all processes are purely natural and unguided, and that phylogenetic history is mainly the product of common descent, at least in multicellular organisms. The four main processes thought to affect population genetics — mutation, genetic drift, gene flow, and selection — are all unguided. The first three are random in their effect on evolution, meaning that they can be positive, negative or neutral in their effects on fitness; only natural selection acts in a directional manner to increase fitness.
There has been debate among population geneticists for decades about the relative importance of these four processes. (For a discussion of this debate go here and here.) The debate is still going on, though with the advent of modern sequence data, the scale has tipped in favor of genetic drift as the predominant process that drives genetic change. This assumption can be seen in Paul McBride’s review of our book, Science and Human Origins.
The theory is that in small populations (smaller than a trillion, say) drift can overwhelm the power of selection. In such a case, organisms do not have sufficient numbers for beneficial mutations to arise and be fixed with any frequency. Most mutations are lost to drift before becoming established, even when they are beneficial. The significance of natural selection is thus greatly reduced in shaping evolutionary history.
The idea that evolution is driven by drift has led to a way of retrospectively estimating past genetic lineages. Called coalescent theory, it is based on one very simple assumption — that the vast majority of mutations are neutral and have no effect on an organism’s survival. (For a review go here.) Under this theory, actual genetic history is presumed not to matter. Our genomes are full of randomly accumulating neutral changes. When generating a genealogy for those changes, their order of appearance doesn’t matter. Trees can be drawn and mutations assigned to them without regard to an evolutionary sequence of genotypes, since genotypes don’t matter.
Here’s the way a recent article put it:
… the genealogical relationship (gene tree) of neutral alleles can be simply depicted by a coalescence process in which lineages randomly coalesce with each other backward in time. The coalescence model is simple in the sense that it assumes little or no effect of evolutionary forces such as selection, recombination, and gene flow, instead giving a prominent role to random genetic drift.
Thus, according to this theory, if it can be assumed that most mutations or allelic states have no effect on fitness, a genealogy can be created randomly without any input from the genotype. Therefore the spread of variation can be modeled as a diffusion process or Markov chain run backwards, the mean time to coalescence can be estimated, and the effective population size can be estimated from that, based on mutation rate and generation time.
Li and Durbin’s paper, cited by Paul McBride in his review, is one such study. The authors use a sequential hidden Markov model, combined with an estimation of historical recombination events, to reconstruct human genetic history. They begin by testing their model on artificial genome sequences they had generated themselves, to see if the algorithm can recover those histories accurately. They also test their algorithm to see the effect of sudden population size change, hotspots of recombination, and variable mutation rate on their ability to reconstruct history, but they test them individually.
The authors then analyze six individual human genomes and retrospectively estimate the times to coalescence and effective population sizes for those individuals. Their analysis is limited to a time span of 20,000 years ago to 3 million years ago, because outside that range, there would be either too few or too many recombination events to be sorted out meaningfully.
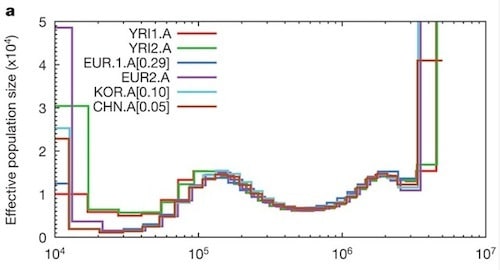
Figure 3a from Li and Durbin. Years (generation time = 25 years, mutation rate = 2.5 x 10-8)
The results of their analysis are summarized in the graph above (this is the same figure McBride included in his post). Each colored line represents the regenerated history from an individual genome. The estimated history does not include the proposed chimp/human split, because of the model’s limitations with regard to very recent and very ancient events. Thus the graph represents the supposed history of the hominin lineage leading to Homo sapiens.
What is clear from the graph is that the model detects several bottlenecks and expansions, with the most recent bottleneck perhaps as small as a thousand individuals, some twenty to sixty thousand years ago. (This bottleneck, they propose, may reflect our emergence from Africa.) As far as modern Homo sapiens‘ first appearance, they place that at some time between sixty and two hundred thousand years ago, with an estimated population size of about fifteen thousand, though they do say that various kinds of population admixtures may have inflated their size estimate.
Here’s my hesitation about retrospective calculations of this kind. Algorithms of this kind report estimates of mean times to coalescence and mean effective population sizes, but they are rarely reported with estimated variance clearly stated. The study I cited in a recent post was an exception in this regard. Li and Durbin’s study had a sample size of six genomes, so any conclusions drawn should be lightly held. In particular, I notice that only one set of data is reported with standard deviations associated. For the smallest bottlenecks, no information on variance or standard deviation is reported.
Second, the complexity of actual history may matter. For example, recent strong bottlenecks followed by rapid exponential growth can wipe out or reduce signal due to earlier events. (See here for a discussion.) Our genomes are much more complicated than the models allow for, or the data can resolve. In particular, strong selection, including balancing selection and overdominant selection can affect estimates of heterozygosity and linkage disequilibrium, which would influence their recovered history. These may lead to erroneous estimates of age for chromosomal segments. Additionally, fluctuating population size, and gene flow between previously isolated populations can lead to overestimates of effective population size.
Coalescent models rely heavily on the assumption that genetic change is neutral. There are indications that this is not the case in our genome as I have already discussed. So conclusions about population size drawn from these models should be provisional at best. In addition, all these calculations depend on assumptions of common descent as the only explanation for our origin. Such assumptions are not justified, when we see evidence of tangled trees at all levels of phylogeny. In an ideal world, such assumptions would have to be justified before being used in models such as this.
The evidence is still coming in. I don’t know how things will turn out in the end. But I suspect the tale of our ancestral population size will be much like the shrinking estimates of our sequence similarity with chimps. The more things are examined in detail, the smaller the numbers will get. It will be interesting to see how small they become.

