 Evolution
Evolution
 Intelligent Design
Intelligent Design
Computing the "Best Case" Probability of Proteins from Actual Data, and Falsifying a Prediction of Darwinism
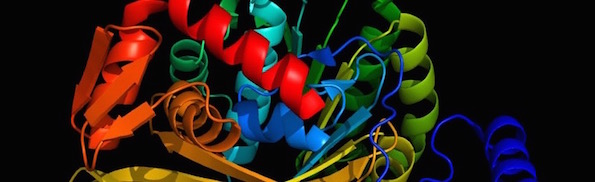
Biological life requires thousands of different protein families, about 70 percent of which are “globular” proteins, with a three-dimensional shape that is unique to each family of proteins. An illustration is shown in the picture at the top of this post. This 3D shape is necessary for a particular biological function and is determined by the sequence of the different amino acids that make up that protein. In other words, it is not biology that determines the shape, but physics. Sequences that produce stable, functional 3D structures are so rare that scientists today do not attempt to find them using random sequence libraries. Instead, they use information they have obtained from reverse-engineering biological proteins to intelligently design artificial proteins.
Indeed, our 21st-century supercomputers are not powerful enough to crunch the variables and locate novel 3D structures. Nonetheless, a foundational prediction of neo-Darwinian theory is that a ploddingly slow evolutionary process consisting of genetic drift, mutations, insertions, and deletions must be able to “find” not just one, but thousands of sequences pre-determined by physics that will have different stable, functional 3D structures. So how does this falsifiable prediction hold up when tested against real data? As ought to be the case in science, I have made my program available so that you can run your own data and verify for yourself the kinds of probabilities these protein families represent.
This program can compute an upper limit for the probability of obtaining a protein family from a wealth of actual data contained in the Pfam database. The first step computes the lower limit for the functional complexity or functional information required to code for a particular protein family, using a method published by Durston et al. This value for I(Ex) can then be plugged into an equation published by Hazen et al. in order to solve the probability M(Ex)/N of “finding” a functional sequence in a single trial.
I downloaded 3,751 aligned sequences for the Ribosomal S7 domain, part of a universal protein essential for all life. When the data was run through the program, it revealed that the lower limit for the amount of functional information required to code for this domain is 332 Fits (Functional Bits). The extreme upper limit for the number of sequences that might be functional for this domain is around 10^92. In a single trial, the probability of obtaining a sequence that would be functional for the Ribosomal S7 domain is 1 chance in 10^100 … and this is only for a 148 amino acid structural domain, much smaller than an average protein.
For another example, I downloaded 4,986 aligned sequences for the ABC-3 family of proteins and ran it through the program. The results indicate that the probability of obtaining, in a single trial, a functional ABC-3 sequence is around 1 chance in 10^128. This method ignores pairwise and higher order relationships within the sequence that would vastly limit the number of functional sequences by many orders of magnitude, reducing the probability even further by many orders of magnitude — so this gives us a best-case estimate.
What are the implications of these results, obtained from actual data, for the fundamental prediction of neo-Darwinian theory mentioned above? If we assume 10^30 life forms with a fast replication rate of 30 minutes and a huge genome with a very high mutation rate over a period of 10 billion years, an extreme upper limit for the total number of mutations for all of life’s history would be around 10^43. Unfortunately, a protein domain such as Ribosomal S7 would require a minimum average of 10^100 trials. In other words, the sum total of mutational events for the entire theoretical history of life falls short by at least 57 orders of magnitude from what would have a reasonable expectation of “finding” any RS7 sequence — and this is only for one domain. Forget about “finding” an average sized protein, not to mention thousands.
As we all know from probabilities, you can get lucky once, but not thousands of times. This definitively falsifies the fundamental prediction of Darwinian theory that evolutionary processes can “find” functional protein families. A theory that has an essential prediction thoroughly falsified by the data should have no place in science.
Could natural selection come to the rescue? As we know from genetic algorithms, an evolutionary “search” will only work for hill-climbing problems, not for “needle in a haystack” problems. There are small proteins that require such low levels of functional information to perform simple binding tasks that they form a nice hill-climbing problem that can be easily located in a search. This is not the case, however, for the vast majority of protein families. As real data shows, the probability of finding a functional sequence for one average protein family is so low, there is virtually zero chance of obtaining it anywhere in this universe over its entire history — never mind finding thousands of protein families.
What are the implications for intelligent design science? A testable, falsifiable hypothesis of intelligent design can be stated as follows:
A unique attribute of an intelligent mind is the ability to produce effects requiring a statistically significant level of functional information.
Given the above testable hypothesis, if we observe an effect that requires a statistically significant level of functional information, we can conclude there is an intelligent mind behind the effect. The average protein family requires a statistically significant level of functional, or prescriptive, information. Therefore, the genomes of life have the fingerprints of an intelligent source all over them.
Image: RecA prepared by Kirk Durston using MacPyMOL.