 Intelligent Design
Intelligent Design
The Finely Tuned Genetic Code
Francis Crick regarded the genetic code found in nature as a “frozen accident.” Yet more and more it is looking to be the case that this code is exquisitely finely tuned — with features suggesting it is indeed one in a million. Therefore ought not purposive or intelligent design be regarded as a legitimate inference, as the best explanation for how the code came into existence?
We are all familiar with the genetic code by virtue of which an mRNA transcript is translated into the amino acid residues that form proteins. Triplets of nucleotides — called “codons” — serve as “molecular words,” each of them specifying a particular amino acid or the stop sites of open reading frames (ORFs). Ribosomes and tRNA-methionine complexes (called “charged” methionyl tRNAs) attach near the 5′ end of the mRNA molecule at the initiation codon AUG (which specifies the amino acid methionine) and begin to translate its ribonucleotide sequences into the specific amino acid sequence necessary to form a functional protein. Each amino acid becomes attached at its carboxyl terminus to the 3′ end of its own species of tRNA by an enzyme known as amino-acyl tRNA synthetase.
Two sites exist on a ribosome for activated tRNAs: the peptidyl site and the amino-acyl site (P site and A site respectively). The initiation codon, carrying methionine, enters the P site. The 3′ UAC 5′ anticodon of the tRNA is paired with the complementary 5′ AUG 3′ mRNA codon. The second tRNA enters the A site. An enzymatic part of the ribosome called peptidyl transferase then creates a peptide bond to link the two amino acids. Upon formation of the peptide bond, the amino-acyl bond that connected the amino acid to its corresponding tRNA is broken, and the tRNA is thus able to leave the P site. This is followed by ribosomal translocation to position a new open codon in the empty A site and also move the second tRNA — which is now bonded to a dipeptide — from the A to the P site. And so the cycle repeats until the occurrence of a stop codon that prevents further chain elongation.
For a visual illustration of how this works in practice, I refer readers to the following short animation:
The total number of possible RNA triplets amounts to 64 different codons. Of those, 61 specify amino acids, with the remaining three (UAG, UAA and UGA) serving as stop codons, which halt the process of protein synthesis. Because there are only twenty different amino acids, some of the codons are redundant. This means that several codons can code for the same amino acid. The cellular pathways and mechanisms that make this 64-to-20 mapping possible is a marvel of molecular logic. It’s enough to make any engineer to drool. But the signs of design extend well beyond the sheer engineering brilliance of the cellular translation apparatus. In this article, I will show several layers of design ingenuity exhibited by this masterpiece of nanotechnology.
How Is the Genetic Code Finely Tuned?
As previously stated, the genetic code is degenerate. This means that multiple codons will often signify the same amino acid. This degeneracy is largely caused by variation in the third position, which is recognized by the nucleotide at the 5′ end of the anticodon (the so-called “wobble” position). The wobble hypothesis states that nucleotides that are present in this position can make interactions that aren’t permitted in the other positions (though it still leaves some interactions that aren’t allowed).
But this arrangement is far from arbitrary. Indeed, the genetic code found in nature is exquisitely tuned to protect the cell from the detrimental effects of substitution mutations. The system is so brilliantly set up that codons differing by only a single base either specify the same amino acid, or an amino acid that is a member of a related chemical group. In other words, the structure of the genetic code is set up to mitigate the effects of errors that might be incorporated during translation (which can occur when a codon is translated by an almost-complementary anti-codon).
For example, the amino acid leucine is specified by six codons. One of them is CUU. Substitution mutations in the 3′ position which change a U to a C, A or G result in the alteration of the codons to ones which also specify leucine: CUC, CUA and CUG respectively. On the other hand, if the C in the 5′ position is substituted for a U, the codon UUU results. This codon specifies phenylalanine, an amino acid which exhibits similar physical and chemical properties to leucine. The fact in need of explaining is thus that codon assignments are ordered in such a way as to minimize ORF degradation. In addition, most codons specify amino acids that possess simple side chains. This decreases the propensity of mutations to produce codons encoding amino acid sequences which are chemically disruptive.
Freeland et al. (2000) show that the genetic code is highly optimized — indeed “the best of all possible codes” — taking into account two parameters: first, the relative likelihood of transitions and transversions; and second, the relative impact of mutation.
The Failed Rejection Problem
Another paper, by Lim and Curran (2001), models the specificity of correct codon-anticodon duplex formation during translation. According to their model, for an incorrect duplex to be rejected by the ribosome, it is necessary for it to have at least one uncompensated hydrogen bond: a criterion which presents difficulties when duplexes have a pair of pyrimidines (i.e. U or C) in the codon’s third position, i.e. the wobble position. Pyrimidine bases are somewhat smaller than purine (G and A) bases and, in the wobble position, can allow certain mismatches in the second position to produce non-Watson-Crick pairs that compensate the missing hydrogen bonds. This results in a mistranslation event because the mismatches in the second position are not properly rejected.
This problem can be circumvented by preventing an anticodon’s pyrimidine in the wobble position from forming a pyrimidine pair. Such a modification entails that a single anticodon that could have recognized four codons is now able to recognize only two. So there will now need to be one tRNA for the pyrimidines of the wobble position and another tRNA for the purines of the wobble position. This explains why 32 codons (those ending with A and G) in the standard genetic code are in “family boxes,” and the other 32 (those ending with C and U) are in “split boxes.” Indeed, the selection of the codon boxes that are “split” is determined by the very same stereochemistry that underlies which of the mismatches in the second position are susceptible to the failed rejection problem. The observed symmetry is thus not arbitrary.
Encrypted Stop Signs
Another astonishing feature of the genetic code is that the sequences of stop codons overlap with those of the codons specifying the most abundant amino acids. This means that the genetic code is set up in such a way to dampen the impact of frameshift mutations. A frameshift mutation occurs as the result of indels (insertions or deletions) of a number of nucleotides that is non-divisible by three. Such an event causes the reading frame to be shifted, resulting in the production and accumulation of misfolded proteins. The earlier on in the sequence that this indel occurs, the greater the alteration of the protein’s amino-acid sequence.
The genetic code is thought of as being comprised of groups of four codons where the first positions are the same for all four (whereas the third can be occupied by any base). When codons code for the same amino acid, they are referred to as a “codon family.” Half of the genetic code is comprised from such codon families. In the codon families designated AAN and AGN (which categorize Asn/Lys and Ser/Arg triplets respectively), the triplets overlap with the UAA and UAG stop codons which terminate translation. These encrypted stop signs help to prevent the accumulation of misfolded proteins.
As Bollenbach et al. (2007) explain,
…stop codons can easily be concealed within a sequence. For example, the UGA stop codon is only one frameshift away from NNU|GAN; the GAN codons encode Asp and Glu, which are very common in protein sequences. Similarly, UAA and UAG can be frameshifted to give NNU|AAN and NNU|AGN (the AAN codons encode Asn or Lys and AGN gives Ser or Arg). Glu, Lys, Asp, Ser, and Arg are relatively common amino acids in the genome, so the probability of a stop codon arising from a misread of a codon from one of these three amino acids is very high. The fact that a stop codon can be “hidden” in this way using a frameshift means that even a signal sequence that happens to include a stop codon (a problem that is bound to arise sooner or later) can be encoded within the protein sequence by using one of the two reading frames in which the stop codon encodes for a frequently used amino acid.
Remarkably, the 64-to-20 mapping system is set up in order to minimize the number of amino acids that are translated from a frameshifted transcript before the appearance of one of the stop codons. Highly frequent codons (e.g. those coding for aspartic or glutamic acid) can frequently form stop codons in the event of a frame shift. Thus, in the conventional genetic code, translation of a frameshift error is halted faster on average than in 99.3% of alternative codes (Itzkovitz and Alon, 2007).
Related to this is the ability, also reported by Itzkovitz and Alon, of the universal genetic code to “allow arbitrary sequences of nucleotides within coding sequences much better than the vast majority of other possible genetic codes.” They report,
We find that the universal genetic code can allow arbitrary sequences of nucleotides within coding regions much better than the vast majority of other possible genetic codes. We further find that the ability to support parallel codes is strongly correlated with an additional property — minimization of the effects of frameshift translation errors.
The genetic code is thus highly optimized for encoding additional information beyond the amino acid sequence in protein-coding sequences. Examples include RNA splicing signals and information about where nucleosomes should be positioned on the DNA, as well as sequences for RNA secondary structure.
Nature’s Alphabet is Non-Random
Philip and Freeland (2011) take this theme to an even deeper level, suggesting that the set of 20 amino acids used in nature is fundamentally non-random. The authors compared the coverage of the standard alphabet of 20 amino acids for “size, charge, and hydrophobicity with equivalent values calculated for a sample of 1 million alternative sets (each also comprising 20 members) drawn randomly from the pool of 50 plausible prebiotic candidates.”
The authors report that,
…the standard alphabet exhibits better coverage (i.e., greater breadth and greater evenness) than any random set for each of size, charge, and hydrophobicity, and for all combinations thereof. In other words, within the boundaries of our assumptions, the full set of 20 genetically encoded amino acids matches our hypothesized adaptive criterion relative to anything that chance could have assembled from what was available prebiotically.
The authors are thus quick to dismiss the chance hypothesis as a non-viable option. The significance of this extends further, for the researchers also go after the eight prebiotically plausible amino acids that are found among the 20 that are currently exhibited in biological proteins. They compared the properties of these amino acids with alternative sets of eight drawn randomly, establishing — once again — the fundamentally non-random nature of those utilized.
The Non-Evolvability of the Genetic Code
Changes in codon assignments would be catastrophic to the cell because such a mutation would ultimately lead to changes to the amino acid sequence in every protein produced by the cell. This means that one cannot have a significantly evolving genetic code, though — it may be granted — there are one or two minor variations on the standard genetic code. Some have tried to argue around this by positing that the lesser-used codons can be redesignated to a different but related amino acid, thus allowing the genetic code to become optimized. There are, however, significant difficulties with this proposal. For one thing, it seems highly unlikely that by virtue of replacing some of the lesser-used amino acid assignments with a related amino acid that one could attain the level of optimization which we find in the conventional code.
Furthermore, the question is naturally raised as to what selective-utility would be exhibited by the new amino acids. Indeed, they would have no utility until incorporated into proteins. But that won’t happen until they are incorporated into the genetic code. And thus they must be synthesized by enzymes that lack them. And let us not forget the necessity for the dedicated tRNAs and activating enzymes which are needed for including them in the code.
One related difficulty with standard evolutionary explanations is that a pool of biotic amino acids substantially less than 20 is liable to substantially reduce the variability of proteins synthesized by the ribosomes. And prebiotic selection is unlikely to sift the variational grist for this trait of amino-acid-optimality prior to the origin of self-replicative life (in many respects, “prebiotic selection” is somewhat oxymoronic).
There is also the added problem of the potential for codon mapping ambiguity. If, say, 80% of the time a particular codon specifies one amino acid and 20% of the time specifies another, this mapping ambiguity would lead to cellular chaos.
For a thorough discursive review of various attempts at explaining code evolution, I refer readers to this 2009 paper by Eugene Koonin and Artem Novozhilov. They conclude their critical review by lamenting that,
In our opinion, despite extensive and, in many cases, elaborate attempts to model code optimization, ingenious theorizing along the lines of the coevolution theory, and considerable experimentation, very little definitive progress has been made.
They further report,
Summarizing the state of the art in the study of the code evolution, we cannot escape considerable skepticism. It seems that the two-pronged fundamental question: “why is the genetic code the way it is and how did it come to be?,” that was asked over 50 years ago, at the dawn of molecular biology, might remain pertinent even in another 50 years. Our consolation is that we cannot think of a more fundamental problem in biology.
Nonetheless, even if we grant the premise that the genetic code can be modified over time, it still remains to be determined whether there are sufficient probabilistic resources at hand to justify appeals to the workings of chance and necessity. In view of the sheer number of codes that would need to be sampled and evaluated, evolutionary scenarios seem quite unlikely.
Doing the Math
Hubert Yockey, a biophysicist and information theorist, has argued that the number of potential genetic codes is of the order of 1.40 x 10^70. Yockey concedes the extremely conservative figure of 6.3 x 10^15 seconds for the time available for the genetic code to evolve. Note that this assumes that the genetic code has been evolving since the Big Bang. So, how many codes per second would be required to be evaluated in order for natural selection to “stumble upon” the universal genetic code found in nature? The math works out to roughly 10^55 codes per second.
Think about that. Even granting such absurd estimates — all the time available since the Big Bang — natural selection would be required to evaluate 10^55 genetic codes per second in order to have a reasonable chance of stumbling across the optimized genetic code found in nature. This treacherous hurdle is accentuated when one considers more reasonable estimates. The earth likely became bio-habitable about 3.85 billion years ago, with signs of the first life appearing around 3.8 billion years ago. More realistic estimates for the time available make the problem only more daunting. For further discussion of this, see biochemist Fazale Rana’s book, The Cell’s Design.
Overlapping Codons and Ribosomal Frameshifting
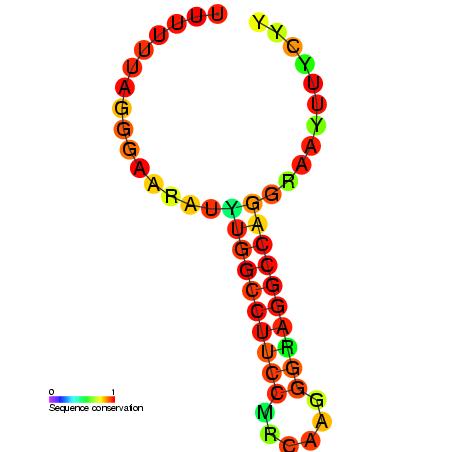 A further remarkable design feature of the genetic code is its ability to have overlapping reading frames such that two or more proteins can be produced from the same transcript. This phenomenon is known as “ribosomal frameshifting”, and is commonly found in viruses including barley yellow dwarf virus, potato leafroll virus and simian retrovirus-1.
A further remarkable design feature of the genetic code is its ability to have overlapping reading frames such that two or more proteins can be produced from the same transcript. This phenomenon is known as “ribosomal frameshifting”, and is commonly found in viruses including barley yellow dwarf virus, potato leafroll virus and simian retrovirus-1.
Ribosomal frameshifting is promoted by a pseudoknot structure (shown in the diagram) and also a specific site in the mRNA, known as a slippery sequence which normally contains several adenine residues. When this occurs, the ribosome shifts back one base and subsequently proceeds to read the mRNA transcript in a different frame. This allows two or more different proteins to be produced from the same transcript!
As mentioned, this programmed ribosomal frameshifting is particularly prevalent in viruses, where the genome must be small because of the small volume of the viral capsid).
It is now known that ribosomal frameshifting occurs in all three of life’s domains. One example of such in eukaryotic cells comes from the yeast Saccaromyces cerevisiae, in which this process produces the proteins Est3p and Abp140p.
Conclusion
In light of facts such as the above, it is becoming increasingly clear that the genome is bidirectional, multifaceted and interleaved at every tie. Unguided chance/necessity mechanisms are demonstrably inadequate in accounting for this engineering marvel. Such delicately balanced and finely tuned parameters are routinely associated with purposive agents. Agents are uniquely endowed with the capacity of foresight, and have the ability to visualize and subsequently actualize a complex end point. If, in every other realm of human experience, such features are routinely associated with intelligent causes — and only intelligent causes — are we not justified in positing that this system also originated at the will of a purposive and conscious agent?