 Evolution
Evolution
 Intelligent Design
Intelligent Design
The Incredible Design of Vertebrate Blood Clotting
Recently, a commenter on the Center for Science and Culture’s Facebook page asked about a paper by the late biochemist Russell F. Doolittle (1931-2019)1 in relation to Michael Behe’s claim, defended in Darwin’s Black Box2 (and in the video below), that the blood clotting cascade is irreducibly complex. Doolittle claims to show “Step-by-step evolution of vertebrate blood coagulation.” However, in a series of three articles I will respond to this claim. First, I will provide a brief description of the blood clotting cascade, for the purpose of bringing readers unfamiliar with the pathway up to speed. Second, I will discuss why vertebrate coagulation is considered to be an irreducibly complex system that poses a significant challenge to evolutionary explanations. Third and finally, I will summarize the key points of the Doolittle paper and offer an evaluation of whether it calls Behe’s argument into question. Since Doolittle has also published a book dealing with this subject3, in which he elaborates on the arguments expressed in the paper in more detail, I will occasionally refer to things said in the book as well.
As we shall see, Doolittle’s proposed scenario does not even touch upon Behe’s thesis, since even in his purported first step in the evolution of vertebrate blood clotting, Doolittle essentially helps himself to all of the components that Behe argued were part of the irreducibly complex core of the system. Thus, Doolittle never even attempts to explain the evolution of the blood clotting system that Behe described as “irreducibly complex.” Doolittle also completely ignores difficulties in adding new factors to the system via unguided evolutionary processes, including the clotting inhibitors that are needed to prevent excessive clot formation, as well as those factors that dismantle clots. This is a glaring omission, since the emergence of the coagulation cascade along the lines proposed by Doolittle would quite probably result in runaway thrombosis, which would entail a significant fitness cost, to say the least. We shall discuss these problems, among others, in the third part of this series.
The Formation of the Platelet Plug
Blood clotting, also known as coagulation, is a complex physiological process that plays a crucial role in maintaining the integrity of the circulatory system. It involves a series of intricate molecular and cellular interactions that result in the formation of a clot at the site of injury within a blood vessel. This process prevents excessive bleeding while promoting wound healing.
Upon injury, platelets adhere to the exposed collagen fibers in the damaged area.4 The platelets then undergo a conformational change and release various substances stored within their granules, which promote further platelet activation and attract more platelets to the site of injury, forming a plug over the hole. Activated platelets bind together, forming aggregates or clumps, reinforcing the platelet plug and creating a temporary seal over the injured area. This seal is a short-term solution, however, and is not strong enough to hold for long. As blood continues to flow through the injured vessel, it can dislodge the loosely adhering platelets. Thus, the formation of this platelet plug (and the injury itself) initiates the coagulation cascade.
The Fibrin Gel
The coagulation pathway involves many different components, which can be difficult to keep track of. For ease of following the description that follows, I recommend referring to the figure below, which depicts aspects of the human coagulation system. An arrow from one component to another indicates that the former activates the latter. A barred line signifies inhibition of one protein by another.
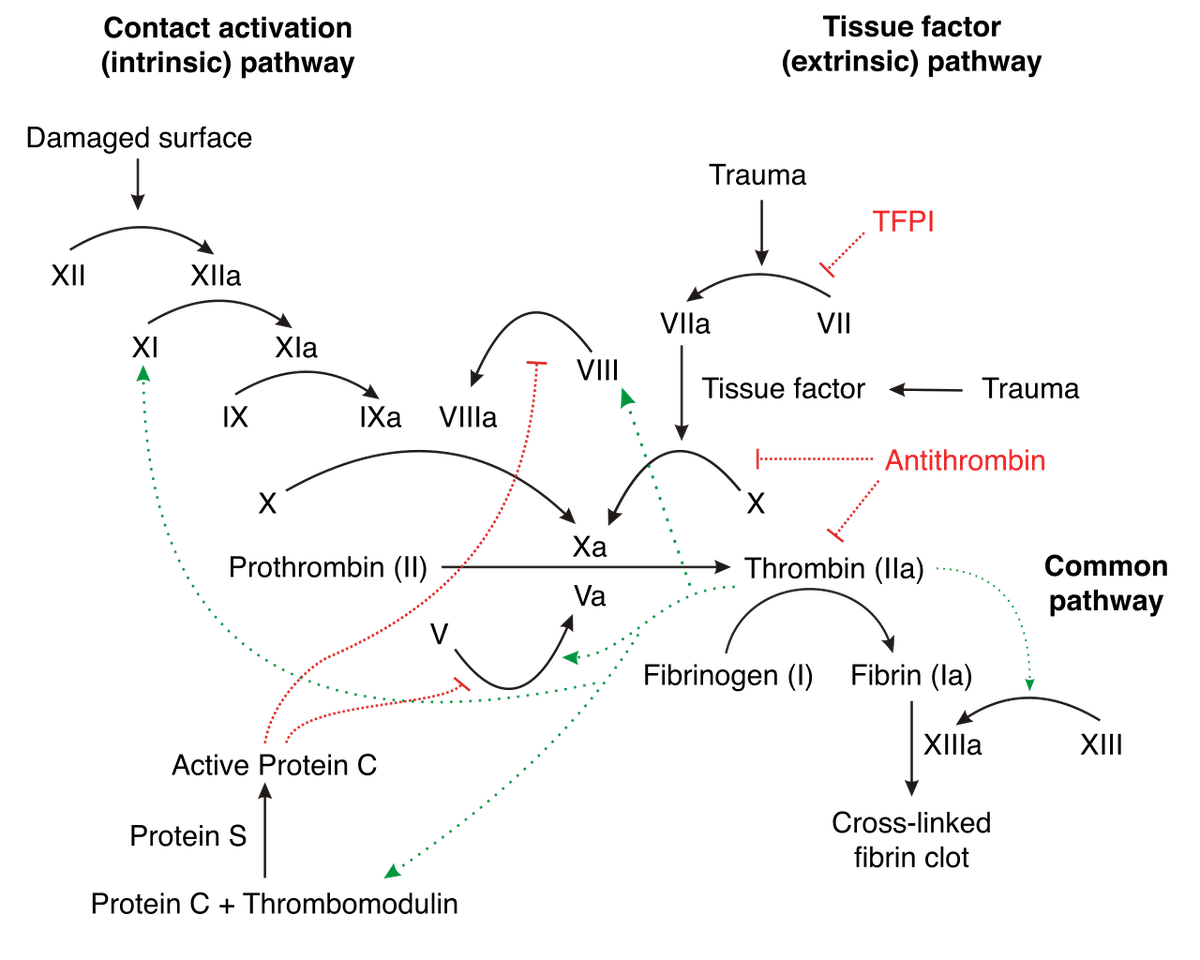
Vertebrate blood coagulation is best understood by focusing first on the ultimate objective of the cascade, which is the formation of a fibrin gel that reinforces the initial platelet plug, thereby strengthening the clot. The clot itself is made of fibers composed of the protein fibrin, which circulates in an inactive form (fibrinogen) in the blood plasma5,6 , shown in the figure below.
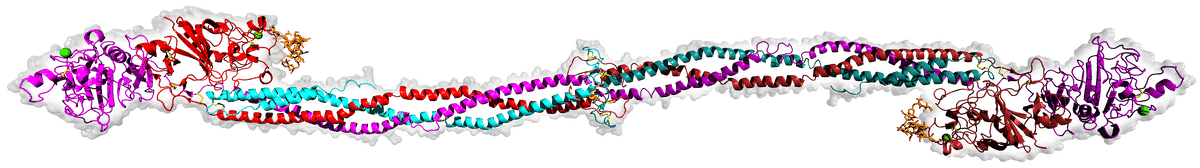
Fibrinogen is comprised of three pairs of polypeptide chains, known as Aα, Bβ, and γ chains, which are held together by disulfide bonds. Fibrinogen becomes activated when another protein, called thrombin, cleaves specific peptide bonds in the fibrinogen molecule, specifically near the N-terminus of the Aα and Bβ chains.7 This cleavage removes small peptide fragments called fibrinopeptides A and B, respectively, from the fibrinogen molecule, resulting in a similar protein with remarkably different functions referred to as fibrin. The cleavage of fibrinopeptides exposes new binding sites on the fibrin molecule, allowing the individual fibrin molecules to polymerize into a clot.8 The fibrin molecules further aggregate and form a mesh-like network, which is stabilized by the enzyme factor XIIIa.9 Factor XIIIa catalyzes the crosslinking of fibrin molecules through the formation of covalent bonds between specific amino acid residues, creating a stable fibrin clot. The resulting fibrin clot, together with the platelets, provides a physical barrier at the site of injury, preventing further blood loss. It also serves as a scaffold for other components of the clotting process, which aggregate on the fibrin network to form a stable blood clot.
If the pathway consisted only of fibrinogen and thrombin, thrombin would constantly cleave fibrinogen, and the consequence would be uncontrolled and excessive clotting throughout the bloodstream. To avoid this, it is essential that the process be carefully regulated. Blood clotting involves the use of proenzymes, which are enzymes that are retained in an inactive state and need to be converted into active enzymes through specific cleavage by proteases such as thrombin.
Thrombin itself exists in an inactive form, prothrombin. To convert prothrombin into active thrombin, another enzyme (factor Xa), along with its cofactor (factor Va), assembles on the surface of platelets or other phospholipid membranes to form the prothrombinase complex.10 This complex provides the platform for the subsequent activation of prothrombin. The conversion of prothrombin to thrombin, mediated by the prothrombinase complex, occurs (as with fibrinogen) through a proteolytic cleavage of prothrombin at specific sites. Curiously, factor V also exists in an inactive state, but is cleaved and activated (to form factor Va) by thrombin itself (a small trace of which circulates in the bloodstream due to a low rate of cleavage of prothrombin by factor Xa).11 Once activated, factor Va plays a critical role in the amplification of the coagulation process by enhancing the activity of factor X and promoting the production of more thrombin. This makes the coagulation cascade autocatalytic, since the activation of clotting factors leads to the activation of more of the same proteins.
Factor Xa also exists in an inactive form, factor X. Factor X may be activated by two different pathways: the extrinsic and intrinsic pathway. In the extrinsic pathway, so-named because it is triggered by the external factors, tissue factor (also known as factor III) is released.12 Tissue factor forms a complex with factor VII, leading to the activation of factor X. The first step of the intrinsic pathway, so-named because all of the components required for its initiation and progression are present within the blood itself, involves the activation of factor XII (also called Hageman factor) by contact with negatively charged surfaces, such as exposed collagen at the site of injury.13 Activated factor XII then activates factor XI, which, in turn, activates factor IX. Activated factor IX (IXa) forms a complex with its cofactor, factor VIIIa, on a phospholipid surface. This complex, along with calcium ions, is called the tenase complex or intrinsic tenase.14,15 The tenase complex plays a crucial role in amplifying the clotting process by cleaving, and thereby activating, factor X.
Preventing Excess Clotting
To prevent excess clotting and ensure that the clotting cascade remains localized to the site of injury, there are several regulatory mechanisms.16 Antithrombin III (ATIII) is a natural anticoagulant that inhibits the activity of thrombin and several other coagulation factors, including factor Xa and factor IXa. It achieves its anticoagulant effects through a mechanism called “serpin” inhibition.17 Serpins (serine protease inhibitors) are a class of proteins that regulate the activity of proteases, including those involved in blood clotting. ATIII binds to Thrombin’s active site, effectively blocking its ability to cleave fibrinogen into fibrin. By inhibiting thrombin, antithrombin III indirectly helps regulate the activation of factor V and thereby prevents excessive clotting.18
Thrombin not only activates factor V but also activates protein C, which, in the presence of its cofactor protein S, inactivates factor Va (activated form of factor V).19 Protein C cleaves factor Va at specific sites, rendering it less active and inhibiting its procoagulant properties. This negative feedback loop helps to limit and regulate the clotting process. Tissue factor pathway inhibitor (TFPI) is a protein that directly inhibits the activity of factor Xa and the factor VIIa-tissue factor complex.20 By inhibiting these factors, TFPI also indirectly prevents excessive activation of factor V and, thereby, of thrombin.
Clot Retraction and Dissolution
After the clot is formed, it undergoes retraction, which involves the contraction of fibrin by platelets within the clot, resulting in the clot becoming denser.21 This process helps to reduce the size of the clot and brings the edges of the wound closer together. Eventually, as the wound heals, the clot needs to be dissolved to restore normal blood flow. Plasmin, a proteolytic enzyme, breaks down the fibrin meshwork into soluble fragments, leading to the dissolution of the clot.22 Plasmin is generated from plasminogen by tissue plasminogen activator (t-PA) or urokinase-type plasminogen activator (u-Pa), among other molecules.
The Role of Vitamin K
Several of the proteins discussed here depend upon vitamin K for their synthesis — these are prothrombin, factors VII, IX, and X, as well as proteins C and S (i.e., the anticoagulant proteins that serve to inhibit excessive clot formation). Vitamin K is essential for the post-translational modification of these clotting factors. Without adequate vitamin K, these proteins cannot undergo the necessary chemical changes, which would impair their ability to function properly in the coagulation process. For this reason, deficiency in vitamin K can lead to a bleeding disorder known as vitamin K deficiency bleeding, or coagulopathy.
Summary
In summary, vertebrate blood clotting is an incredible, tightly regulated, multi-component cascade that intuitively points to intelligent design. This is in view of its goal-directedness towards its final end, which is producing a successful clot. In a second article in this series, I will discuss why unguided evolutionary explanations are implausible in accounting for the origins of the coagulation pathway.
Notes
- Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35-40.
- Behe, MJ. Darwin’s Black Box: The Biochemical Challenge to Evolution. Free Press 1996.
- Doolittle RF. The Evolution of Vertebrate Blood Clotting. University Science Books 2013.
- Periayah MH, Halim AS, Mat Saad AZ. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int J Hematol Oncol Stem Cell Res. 2017 Oct 1;11(4):319-327.
- Kattula S, Byrnes JR, Wolberg AS. Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arterioscler Thromb Vasc Biol. 2017 Mar;37(3):e13-e21.
- Pieters M, Wolberg AS. Fibrinogen and fibrin: An illustrated review. Res Pract Thromb Haemost. 2019 Mar 4;3(2):161-172.
- Greenberg CS, Miraglia CC, Rickles FR, Shuman MA. Cleavage of blood coagulation factor XIII and fibrinogen by thrombin during in vitro clotting. J Clin Invest. 1985 May;75(5):1463-70.
- Weisel JW, Litvinov RI. Fibrin Formation, Structure and Properties. Subcell Biochem. 2017;82:405-456. doi: 10.1007/978-3-319-49674-0_13.
- Lorand L. Factor XIII and the clotting of fibrinogen: from basic research to medicine. J Thromb Haemost. 2005 Jul;3(7):1337-48.
- Krishnaswamy S. The transition of prothrombin to thrombin. J Thromb Haemost. 2013 Jun;11 Suppl 1(0 1):265-76.
- Keller FG, Ortel TL, Quinn-Allen MA, Kane WH. Thrombin-catalyzed activation of recombinant human factor V. Biochemistry. 1995 Mar 28;34(12):4118-24.
- Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007 Aug;27(8):1687-93.
- Grover SP, Mackman N. Intrinsic Pathway of Coagulation and Thrombosis. Arterioscler Thromb Vasc Biol. 2019 Mar;39(3):331-338.
- Autin L, Miteva MA, Lee WH, Mertens K, Radtke KP, Villoutreix BO. Molecular models of the procoagulant factor VIIIa-factor IXa complex. J Thromb Haemost. 2005 Sep;3(9):2044-56.
- Childers KC, Peters SC, Lollar P, Spencer HT, Doering CB, Spiegel PC. SAXS analysis of the intrinsic tenase complex bound to a lipid nanodisc highlights intermolecular contacts between factors VIIIa/IXa. Blood Adv. 2022 Jun 14;6(11):3240-3254.
- Dahlbäck B. Blood coagulation and its regulation by anticoagulant pathways: genetic pathogenesis of bleeding and thrombotic diseases. J Intern Med. 2005 Mar;257(3):209-23.
- Sanrattana W, Maas C, de Maat S. SERPINs-From Trap to Treatment. Front Med (Lausanne). 2019 Feb 12;6:25.
- van Boven HH, Lane DA. Antithrombin and its inherited deficiency states. Semin Hematol. 1997 Jul;34(3):188-204.
- Kalafatis M, Mann KG. Role of the membrane in the inactivation of factor Va by activated protein C. J Biol Chem. 1993 Dec 25;268(36):27246-57. PMID: 8262965.
- Wood JP, Ellery PE, Maroney SA, Mast AE. Biology of tissue factor pathway inhibitor. Blood. 2014 May 8;123(19):2934-43.
- Tucker KL, Sage T, Gibbins JM. Clot retraction. Methods Mol Biol. 2012;788:101-7.
- Alkjaersig N, Fletcher AP, Sherry S. The mechanism of clot dissolution by plasmin. J Clin Invest. 1959 Jul;38(7):1086-95.