 Evolution
Evolution
Polar Bear Seminar: The APOB Gene and Damaging Mutations


Editor’s note: This is the second installment of a five-part series taking a closer look at Michael Behe’s arguments that polar bear genes experienced adaptive mutations that were damaging. Find the full seminar here.
There is an old joke that goes like this:
Question: When does “damaging” not mean “damaging”?
Answer: When a peer-reviewed paper in a top scientific journal says over 40 times that mutations are predicted to be “damaging,” but then an ID guy comes along and cites the paper to suggest the mutations are probably damaging.
Actually that’s not an old joke, but maybe someday it will be. Anyway, it perfectly describes what is going on right now with critics of Michael Behe and his argument about mutations in polar bear genes.
In Darwin Devolves, Behe cites a paper in the journal Cell, Liu et al. (2014). He does so as evidence that the adaptive mutations in the polar bear gene APOB were “very likely to be damaging — that is, likely to degrade or destroy the function of the protein” (pp. 16-17). Two ID critics, biologists Nathan Lents and Arthur Hunt, object that Behe is wrong because only “some” but “[d]efinitely not all of them or even most of” the mutations in the gene were probably damaging. They go even further and claim that “the authors [of Liu et al. (2014)] do not expect the polar bear APOB to be broken or damaged” and “There is no evidence for Behe’s claim that APOB is degraded or diminished in polar bears.”
No Evidence?
The objection is easy to answer. Behe interpreted the paper correctly and followed its methodology, whereas Lents and Hunt did not. Liu et al. found five specific mutations in APOB that they predicted were important to the gene in allowing polar bears to adapt and shift to a diet higher in fatty acids. The paper says:
In contrast with brown bear, which has no fixed APOB mutations compared to the giant panda genome, we find nine fixed missense mutations in the polar bear. Five of the nine cluster within the N-terminal ba1 domain of the APOB gene, although the region comprises only 22% of the protein (binomial test p value = 0.029). This domain encodes the surface region and contains the majority of functional domains for lipid transport. We suggest that the shift to a diet consisting predominantly of fatty acids in polar bears induced adaptive changes in APOB, which enabled the species to cope with high fatty acid intake by contributing to the effective clearance of cholesterol from the blood.
The diagram below shows all the data extracted from Table S7 in the paper that pertains to mutations in the gene APOB (note: the full Table S7 is presented at this end of this post):
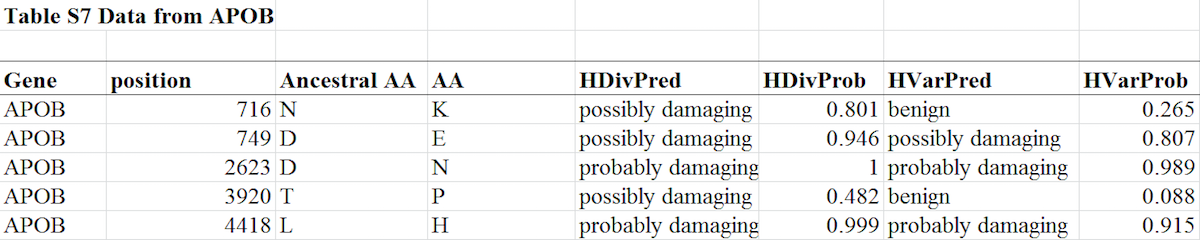
As you can see, the paper reports the results of two algorithms (HDiv and Hvar), and for all five of the APOB mutations at least one algorithm predicted it was damaging. Some of the APOB mutations are said to be “possibly damaging” while others are “probably damaging.” And in two cases, one of the algorithms predicted the mutation was “benign,” while the other predicted it was “possibly damaging.” What does this mean, and how do the authors of the paper themselves interpret this data?
To assess whether each mutation was likely to be damaging to the protein’s function, the researchers used a program called PolyPhen-2. The program works by taking a gene from an organism and comparing it to homologous versions in humans and other organisms. It then looks at the amino acid differences in the version of the gene that’s being studied compared to the homologues, and predicts whether those changes were benign or damaging to the protein (more on what this means below). Using complex algorithms, the program then provides scores that show the likelihood that the mutation is damaging to the protein’s function. Two different algorithms are used and each gives a prediction.
Probabilistic Results
The program offers probabilistic, not absolute, results. This is fully consistent with Behe’s description of the paper’s findings in probabilistic terms, stating that the mutations were “very likely to be damaging — that is, likely to degrade or destroy the function of the protein.”
It’s also important to note that in the PolyPhen-2 program, whether a mutation is labeled “probably damaging” or “possibly damaging,” in either case the evidence evaluated by the program indicated the protein’s function was probably damaged. The reason the program sometimes labels a mutation “possibly” instead of “probably” damaging is not because the evidence was necessarily weak (or less than 50 percent probability). Instead, the label “possibly” was assigned out of concern for potential false positives. But even in those cases when the program says “possibly damaging,” good evidence points towards its being damaging. Just look at how Liu et al. (2014) interpreted the results of the PolyPhen-2 algorithms: In Figure 4C of the paper and in the paper’s text, it’s clear that mutations that are “probably” or “possibly” damaging are said to be “functionally damaging” to the protein. Here’s how the paper puts its findings:
[W]e assessed the impact of polar bear — specific substitutions on human proteins for top-20 genes under positive selection by computational predictions: a large proportion (ca. 50%) of mutations were predicted to be functionally damaging (Figures 4C and 4D, Table S7). (Emphasis added.)
How did the paper come up with the “ca. 50%” statistic? Looking at the full Table S7 (see below), 48 total mutations were studied, and for 25 of those mutations (25/48 = 52 percent), at least one of the methodologies predicted the mutation was either “possibly” or “probably” damaging. And note: under the methodology of the paper, a mutation qualifies as damaging even if just one of the algorithms predicted it as being so. Thus, in the passaged quoted above, the paper identified both categories of mutations as having been “predicted to be functionally damaging.”
When he claimed that a given mutation was predicted to be damaging, Behe simply followed the methodology of Liu et al. In the paper, if one of the algorithms predicted the mutation was “possibly” or “probably” damaging to the gene, then it is said to be “functionally damaging.” Behe was correct in saying that the adaptive mutations in APOB “were very likely to be damaging.” And note that this pertains to all of the five mutations listed for APOB in Table S7, not merely “some” nor “Definitely not all of them or even most,” as Lents and Hunt claim.
You Might Be Wondering
At this point you might be wondering how Lents and Hunt got their idea that Behe wrongly claimed the mutations in this gene are likely damaging. As we saw, Table S7 plainly states that each of the five adaptive mutations in this gene is predicted to be damaging.
They don’t do a very good job of explaining their thinking, but to say that less than half of the APOB mutation should be considered damaging, it seems Lents and Hunt consider a mutation damaging if and only if both algorithms predict it was “probably damaging.” This counting method differs markedly from the methodology of Liu et al. How so?
- The paper considered a mutation to be “predicted to be functionally damaging” if an algorithm found it was “probably” or “possibly” damaging, but Lents and Hunt exclude any mutation where an algorithm found it was “possibly” damaging.
- The paper considered a mutation to be “predicted to be functionally damaging” even if only one algorithm predicted it was damaging, but Lents and Hunt require both algorithms to find it is damaging in order to call it “damaging.”
You could argue all day about the best way to evaluate the data. The bottom line is that this peer-reviewed paper in Cell used a methodology that qualifies all five mutations in APOB as “predicted to be functionally damaging.” Behe followed the paper’s methodology; Lents and Hunt did not.
Let’s Try Again
Lents and Hunt try another argument:
Clearly, the authors do not expect the polar bear APOB to be broken or damaged. Rather, a bare majority of the amino acid changes are clustered in the most important region for the clearing of cholesterol from the blood. This argues that these mutations likely enhance the function of apoB, at least when it comes to surviving on a diet high in saturated fats.
It is also worth noting that apoB does much more than clear fatty acids from the blood. It is a very large protein that has many biochemical activities and is a central player for lipid and cholesterol transport. Even if “damaging” mutations might be beneficial in one context, they could very well be harmful or lethal in another. Moreover, mice that lack apoB are not viable.
To recap: 1.) There is no evidence for Behe’s claim that APOB is degraded or diminished in polar bears and everything we know about the protein from other mammals suggests the opposite. (Emphasis added.)

They say that the authors “do not expect the polar bear APOB to be broken or damaged.” Yet the paper directly contradicts their interpretation. Eight analyses of mutations in apoB listed in Table S7 reported those mutations as “possibly damaging” or “probably damaging” to the protein, and in the text of the paper, the authors included these mutations in APOB among those that were “were predicted to be functionally damaging.”
When Lents and Hunt claim that the mutations in APOB “likely enhance the function of apoB,” that is their own speculation. They focus on a statement in the paper that the adaptive changes in APOB “enabled the species to cope with high fatty acid intake by contributing to the effective clearance of cholesterol from the blood.” Perhaps that is true, but the paper presents no evidence that the removal of cholesterol was accomplished by constructive mutations. Indeed, as Behe pointed out, cholesterol may be removed by decreasing the activity of APOB.
All of the actual evidence presented by the paper suggests that the adaptive mutations in APOB were degradative. Again, according to the paper, eight analyses of mutations in APOB found that those mutations “were predicted to be functionally damaging.”
Lents and Hunt trumpet the important evidence they believe that Behe left out. Ironically, however, their response never quotes from the paper where it says that mutations including those in APOB “were predicted to be functionally damaging.” Their critique — “There is no evidence for Behe’s claim that APOB is degraded or diminished in polar bears” — is simply false and directly contradicts Liu et al. Indeed, one of the mutations in APOB got the highest possible score — 1.0 — for predicting damage to the protein!
Does Positively Selected Mean Constructive?
At this point you might ask how a damaged gene could be positively selected yet have degradative or constructive mutations. Behe explains in his book:
It’s been known for a long time that the great majority of mutations that have a measurable effect on a creature’s welfare are harmful. The amazing but in retrospect unsurprising fact established by the diligent work of many investigators in laboratory evolution over decades is that the great majority of even beneficial positively selected mutations damage an organism’s genetic information — either degrading or outright destroying functional coded elements.
Why is that the case? The simple reason is that the targets for damaging mutations are just much more numerous than those for gain-of-FCT mutations, so they’ll be hit much more frequently. Suppose a beneficial effect could be obtained by breaking or degrading a gene. A modestly sized gene might consist of upwards of a thousand nucleotides. The ways one could break such a juicy target are legion. … Contrast this with a gene in which one of just a few nucleotides has to be mutated to yield a beneficial effect. That would almost always be the case for a new gain-of-FCT feature (such as a new protein-binding site or posttranslational modification site) because … specific new functional features have to have specific structures in specific places. In other words, there are expected to be far fewer positions in a gene that can be changed to yield a helpful gain-of-FCT. Since mutations occur randomly, and since there are thousands of ways to break or degrade a gene, but perhaps just a handful to improve it constructively, the rate of appearance of a beneficial mutation that breaks or degrades a gene is expected to be hundreds to thousands of times faster than a beneficial mutation that has to change a specific nucleotide in a gene. … Thus damaging mutations will almost always occur first and so have the first opportunity, well before constructive mutations, to be positively selected if they are helpful. (Darwin Devolves, pp. 183-185)
When “Damaging” Doesn’t Mean “Damaging”
Some of Behe’s critics take a different tack. Aware that Liu et al. (2014) states that many mutations in APOB and other polar bear genes “were predicted to be functionally damaging,” they try to redefine the word “damaging.” This was the approach adopted by Joshua Swamidass in a webinar last month. He argued there that “damaging in this table does not mean damaging” because all the Polyphen-2 program is really detecting is a “change in function.” Under this view, despite the fact that the word “damaging” appears over 40 times in the supplemental material published by Liu et al., and despite the fact that the paper itself states that about 50 percent of the mutations studied “were predicted to be functionally damaging,” Michael Behe is mistaken in reporting that any of the mutations were actually functionally damaging. As we’ll see, this view does not adequately or fairly explain what the program is really finding.
Richard Lenski elaborated on this objection at his blog:
Liu et al. analyzed the polar-bear version of the APOB gene using the PolyPhen-2 computational tool described above. Roughly half the mutations in APOB were categorized by that program as “possibly damaging” or “probably damaging,” and the rest were called “benign.” Behe than concluded that some of the mutations had damaged the protein’s function, and that these mutations were beneficial in the environment where the polar bear now lives. In other words, Behe took this output as strong support for his rule.
So what’s the problem? The PolyPhen-2 program, as I explained, is designed to identify mutations that are likely to affect a protein’s structure and therefore its function. It assumes such mutations damage (rather than improve) a protein’s function because structurally similar mutations are rare in humans and other species used for comparison. It does so because it presumes that natural selection has optimized the protein to perform a specific function that is the same in all cases, so that changes must be either benign or damaging to the protein’s function. In fact, the only possible categorical outputs of the program are benign, possibly damaging, and probably damaging. The program simply cannot detect or suggest that a protein might have some improved activity or altered function. (Emphasis in the original.)
Lenski makes some reasonable points here, as Behe recognizes, but nothing he says undermines Behe’s arguments. Note Lenski’s words that the program “assumes such mutations damage (rather than improve) a protein’s function because structurally similar mutations are rare in humans and other species used for comparison.” Actually, programs don’t assume anything. Programmers who write programs build assumptions into their programs, and they often have very good reasons for doing so.
In this case, the program is designed to look at the substituted amino acids and assess whether they have similar chemical properties to the residues at the same position in other homologues. If they don’t, it’s safe to assume that the function of the protein is going to begin to diverge from the original function. That means the protein’s native function is probably being degraded. Thus, in a very large proportion of cases, it’s safe to assume that a mutation diverging from the chemical properties of a suite of homologues will probably damage that protein’s function. That’s exactly what the program looks for. So Swamidass may be correct that the program detects a “change in function,” but the methods used to detect such changes make it very likely that the changes will be damaging.
A Variety of Criteria
According to the PolyPhen-2 website, the program uses a variety of criteria to predict whether the amino acid change was “damaging” or “benign.” The criteria include assessing whether the particular mutation is known to cause diseases in humans, the degree to which other versions of the gene are known to tolerate the mutation, and whether the chemical properties of the new amino acid make it “likely to destroy the hydrophobic core of a protein, electrostatic interactions, interactions with ligands or other important features of a protein.” Other literature on the PolyPhen-2 program provides similar descriptions.
One paper says PolyPhen-2 compares the observed mutations to mutations in homologous genes known to cause “human Mendelian diseases and affecting protein stability or function.” Looking for correlations with disease and assessing protein stability and function means that PolyPhen-2 should be able to directly predict when there has probably been real degradation of the protein’s ability to carry out its native function.
Another paper about PolyPhen-2 explains that it compares sequences that are “damaging alleles with known effects on the molecular function” and further notes that the program:
predicts the possible impact of amino acid substitutions on the stability and function of human proteins using structural and comparative evolutionary considerations.
According to this paper, the program provides “a probability score for the variant to have a damaging effect on the protein function.” Again, the program isn’t just blindly reporting differences in the protein. Instead, it smartly compares the differences it finds to known examples where there are “damaging alleles with known effects on the molecular function” or effects on the “stability” of the protein. These could be very good probabilistic indicators of whether the protein’s function has been degraded.
Yet another paper explains that the program “predicts the effect of an nsSNP [non-synonymous single nucleotide polymorphism] on protein structure and function.”
The point is that the program is potentially capable of analyzing mutations in ways that probably would, or are already known to, degrade a protein’s native function. Lenski himself spells out the logic: If a protein is already optimized for some native function, and a substituted amino acid has chemical properties that make it likely to change that function, then the protein’s function will probably be damaged. That is precisely the way that Behe means “damaged.” As he writes:
They [Liu et al. (2014)] determined that the mutations were very likely to be damaging — that is, likely to degrade or destroy the function of the protein that the gene codes for. (Darwin Devolves, p. 17)
Damaging the function in this way is exactly how Liu et al. define “damaging” when they note that “a large proportion (ca. 50%) of mutations were predicted to be functionally damaging” (emphasis added).
Behe has replied to Lenski. He points out that while Lenski is correct that “computer-assignment of a mutation as ‘damaging’ is not a guarantee,” since you can never rule out the possibility “that the protein may have secretly gained some positive new function,” it is nonetheless the case that algorithmic investigations like that used by PolyPhen-2 can generally be very useful to predict degradative mutations:
The caveats mentioned above by Professor Lenski — about how computer-assignment of a mutation as “damaging” is not a guarantee, and that the protein may have secretly gained some positive new function — are correct. He is also quite right to say that without detailed biochemical and other experiments we cannot know for sure how the change affected the protein and the larger system at the molecular level. Nonetheless, computer methods of analyzing mutations are widely used because they are generally accurate. And they do not suddenly lose their accuracy when I cite their results. So, in the absence of specific information otherwise, that’s the way for a disinterested scientist to bet. There is no positive reason — other than an attempt to fend off criticism of the Darwinian mechanism — to doubt the conclusion.
Remember, the creators of the PolyPhen-2 program deliberately chose to have it output language that says that a mutation is “damaging” — NOT merely “changing” — the function of the protein.
In the end, the PolyPhen-2 program, Liu et al. (2014), and Behe’s description of their findings are all correct and consistent. PolyPhen-2 does not give absolute answers but probabilistic ones. Probabilities, being what they are, may be wrong. But based upon what we know about how proteins work, about what kinds of roles particular amino acids usually play in proteins, and empirical data about how homologous proteins behave when they have specific amino acid changes, the program gives a legitimate estimate of whether a given mutation is going to damage the protein’s native function. Behe has appropriately and accurately described the findings of the program when he writes that it found “the mutations were very likely to be damaging—that is, likely to degrade or destroy the function of the protein that the gene codes for.” (emphases added)
Note that Behe does not say they “were absolutely damaging” or “absolutely degrade or destroy function.” No, Behe says “very likely to be damaging” or “likely to degrade or destroy function.” With his qualifications Behe has appropriately and accurately described the findings of the program: Even if we can’t absolutely rule out the possibility that the mutations are non-damaging, we have a good probabilistic estimate that, as Behe correctly says, it is “very likely” that the mutation degrades the protein’s function.
Science never gives absolute certainty. Direct empirical studies of these mutations in polar bear genes could potentially shed more light on their specific effects. Perhaps that work will support Behe’s thesis, or perhaps it won’t. In the meantime, the best evidence we have shows that Behe is fully justified in drawing his conclusion that the polar bear mutations were probably damaging.
Lastly, here is the full Table S7 from Liu et al. (2014), without edits. Keep reading this series to see why none of the data in the table contradicts Behe’s thesis:
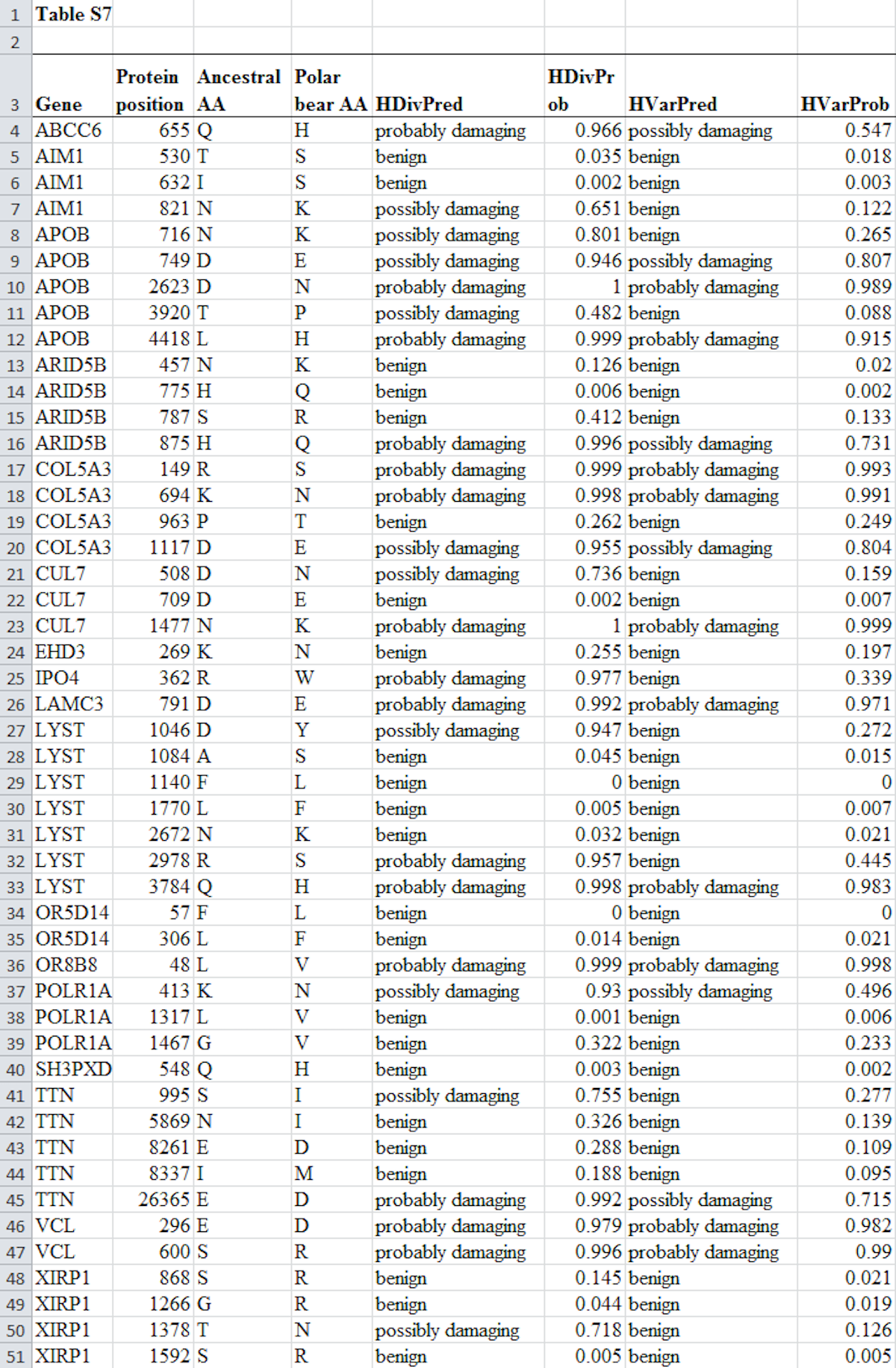
Photo credit: Jacqueline Godany via Unsplash.
Update: Joshua Swamidass now writes in response to this post “Are they just ignoring the fact that the authors concluded that the mutations were not damaging?” This is a false accusation because the authors of Liu et al. (2014) did not conclude that the mutations were not damaging. Rather, as we have repeatedly documented throughout this post, they concluded, “[W]e assessed the impact of polar bear — specific substitutions on human proteins for top-20 genes under positive selection by computational predictions: a large proportion (ca. 50%) of mutations were predicted to be functionally damaging.” (Emphasis added.) That was their conclusion. They concluded that the mutations were “predicted to be functionally damaging.” There is no language to the contrary in the paper, and the burden is upon critics of Behe to provide language which shows that the authors specifically argued that the mutations were not damaging.